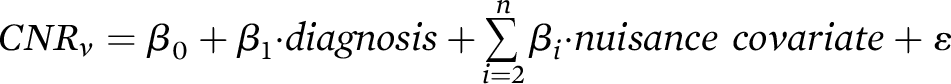Alterations of dopamine function have been demonstrated in cocaine use disorder using positron emission tomography (PET), including measures of dopamine uptake, receptor density, and dopamine release (
1). The reduction of stimulant-induced presynaptic dopamine release in cocaine users, measured with PET, is well replicated (
1–
4) and is associated with more refractory symptoms of cocaine use disorder, including relapse (
1,
2). However, while PET can provide important insights on dopamine signaling in addiction, it is costly and requires significant specialized infrastructure. Furthermore, its use in longitudinal studies and research in younger, at-risk populations is limited by the risks of radioactivity exposure.
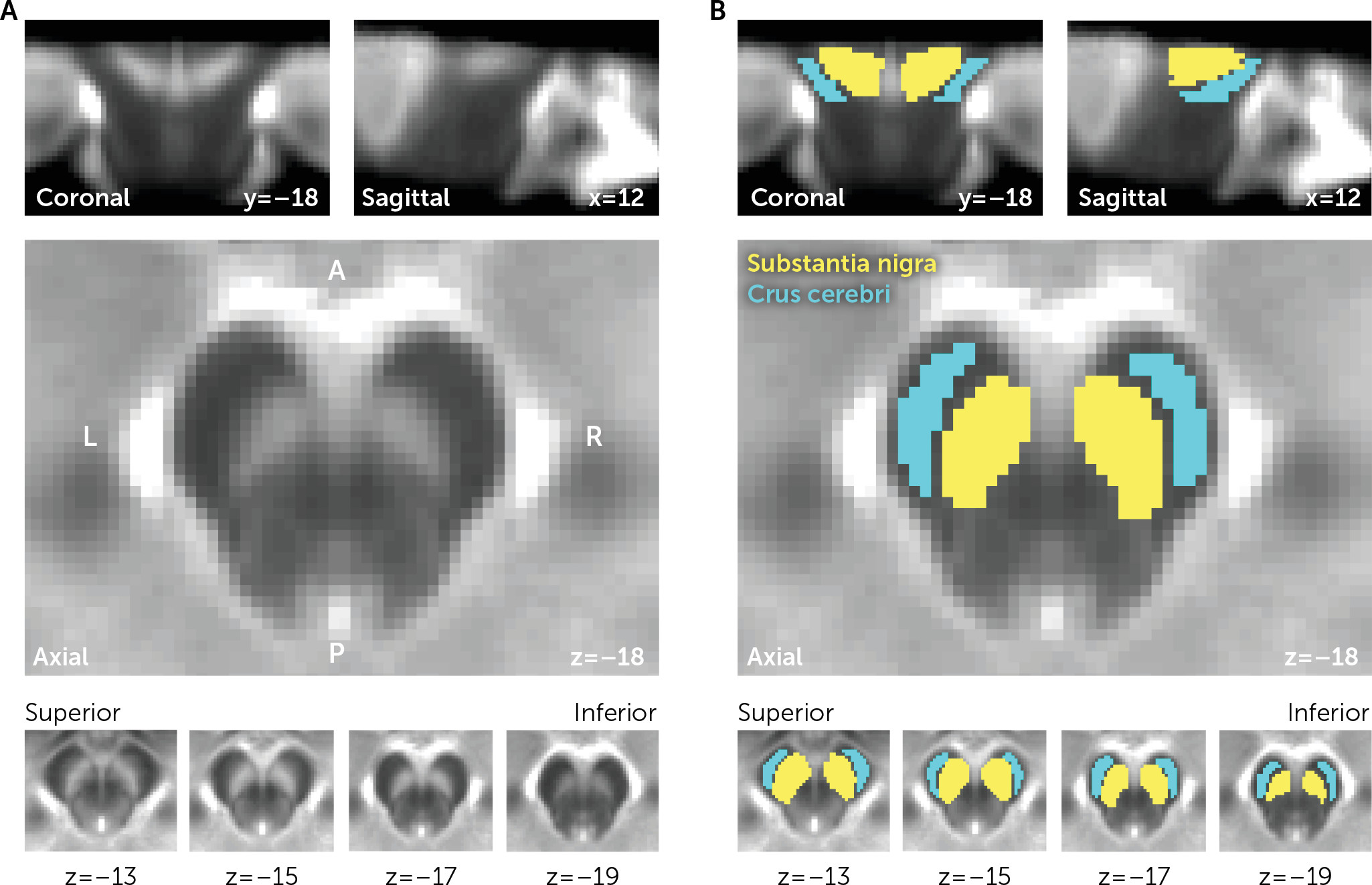
Recent work suggests that neuromelanin-sensitive MRI (NM-MRI) may provide a complementary noninvasive proxy measure of dopamine function and integrity (
5,
6). Neuromelanin (NM) is a pigment generated from the conversion of cytosolic dopamine that accumulates gradually over the lifespan in dopamine neurons of the substantia nigra (SN) (
7). NM is bound to iron, forming paramagnetic complexes that can be imaged using MRI (
6,
8,
9). NM-MRI can reliably capture NM depletion following SN neurodegeneration in Parkinson’s disease (
6,
10). Critically, this technique can also capture alterations in dopamine function in the absence of neurodegeneration (
5,
11), consistent with in vitro evidence that stimulating dopamine synthesis boosts NM synthesis (
12,
13).
In particular, our recent work showed that NM-MRI signal within a subregion of the SN is increased in relation to psychosis (
5), consistent with PET findings of increased dopamine signaling in psychosis (
14). We further showed that NM-MRI signal correlates directly with both PET measures of presynaptic dopamine release and resting blood flow in the midbrain (
5). Thus, this work demonstrates that NM-MRI provides a proxy measure for functional changes in dopaminergic pathways, with utility for studying psychiatric disorders in the absence of overt neurodegeneration.
Here, we sought to employ NM-MRI to examine whether similar changes could be detected in cocaine use disorder, a disorder involving dopamine dysfunction. To this end, our main analyses tested for effects of diagnostic group (individuals with and without cocaine use disorder) on NM-MRI signal in the SN. Our hypothesis, based on previous PET studies (
1,
3), was that cocaine use disorder would be associated with reduced NM-MRI signal. In exploratory analyses, we then evaluated associations between changes in NM-MRI signal intensity in cocaine use disorder and hemodynamic brain responses during the monetary incentive delay task. Activation of the ventral striatum during the anticipation of reward in this task has been shown to provide a robust functional readout of reward processing (
15) related to dopamine (
16,
17) that is consistently reduced in drug and behavioral addictions (
18,
19). Since the ventral striatum receives projections from the ventral tegmental area and the dorsomedial SN (
20,
21), we explored the relationship between NM-MRI signal in the SN and reward-related activation in the ventral striatum.
Discussion
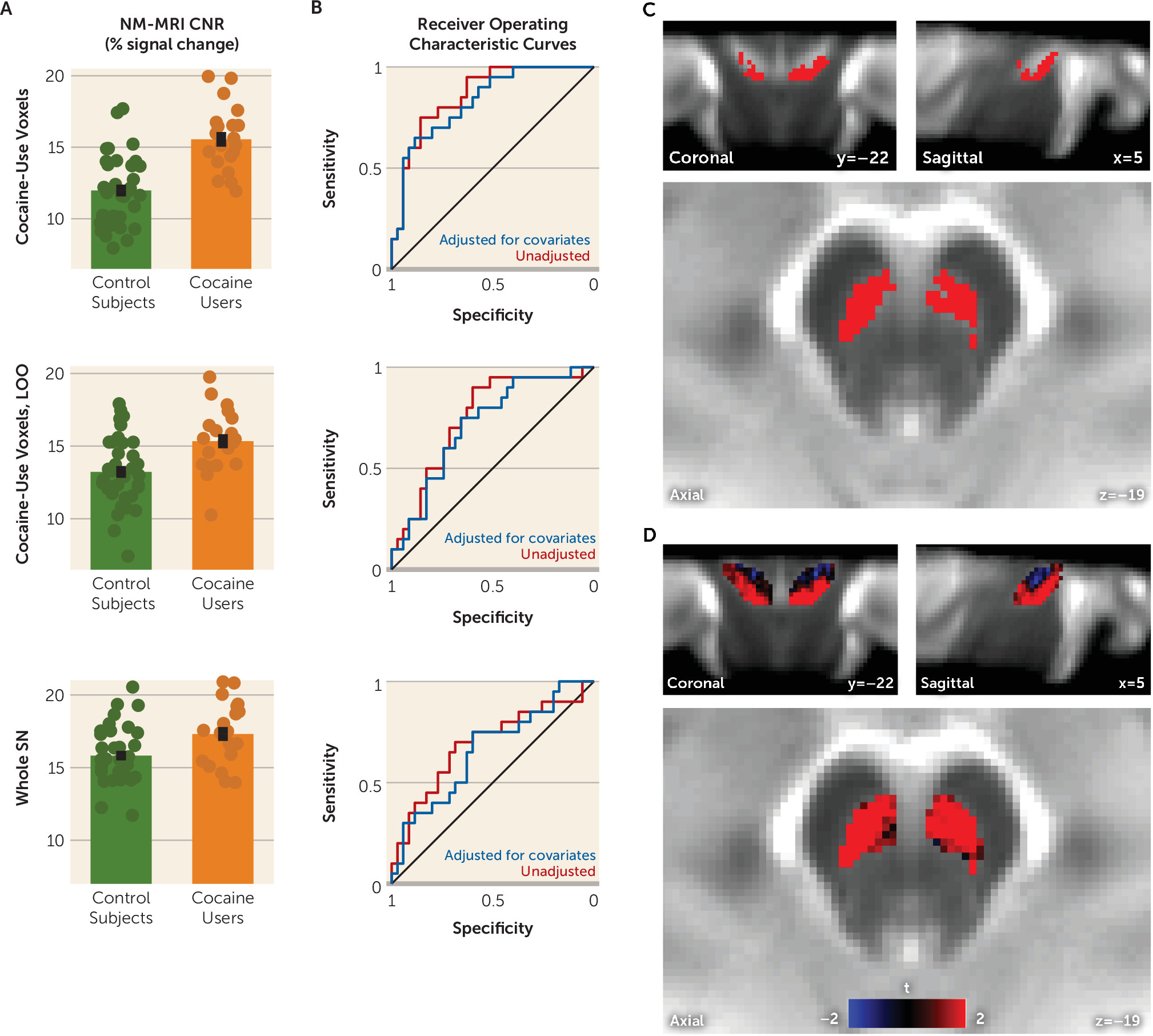
We have presented data showing increased NM-MRI signal in the SN of individuals with cocaine use disorder. This increase was not present throughout the whole SN but rather predominated in more ventral and lateral SN subregions. Given that the NM-MRI signal reflects the concentration of synthetic melanins in experimental preparations (
8) and of NM in postmortem midbrain tissue (
5), and that NM accumulation in the SN depends on dopamine function (
5,
12,
13), these findings suggest that cocaine users exhibit elevated NM concentration in these SN subregions that may be indicative of dopaminergic dysfunction.
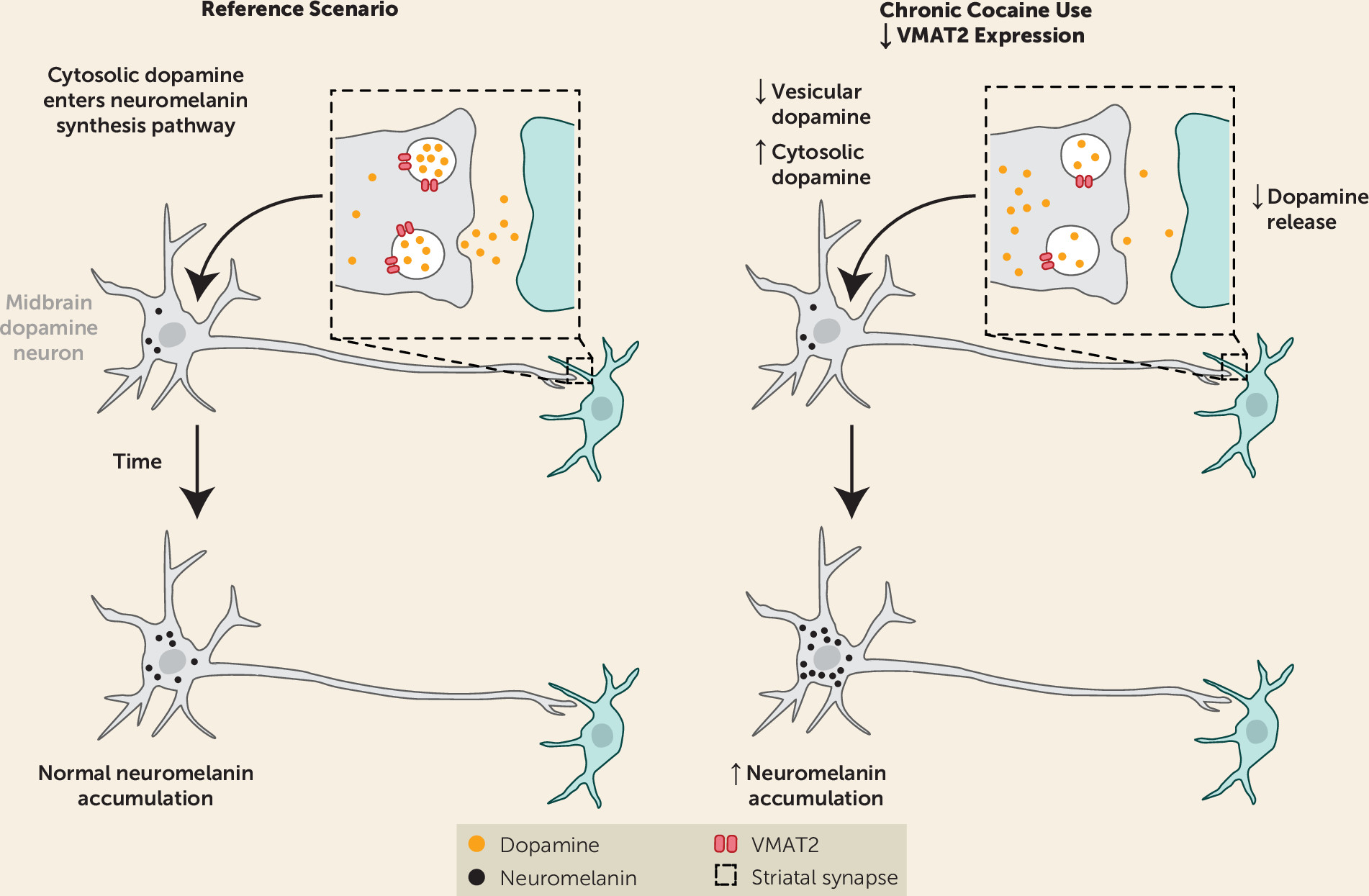
Our finding of elevated NM signal in cocaine users was surprising given the previous PET studies showing that presynaptic dopamine is blunted in cocaine use disorder (
1–
4). However, this discrepancy provides additional insight into the pathophysiology of dopamine signaling in this disorder. The combination of blunted dopamine release in the striatum and elevated NM in the SN suggests that dopamine is distributed differently in cocaine users compared with control subjects. Less dopamine concentrated in synaptic vesicles and more dopamine in the cytosolic pool would explain the divergence between PET studies, which estimate dopamine release from vesicles, and imaging of NM, which accumulates on the basis of the concentration of dopamine in the cytosol (
12,
26). If, on the other hand, cocaine use disorder were associated with a global and persistent decrease in dopamine synthesis, we would expect to have seen a decrease in both the PET and NM-MRI signals.
A number of previous studies support our hypothesis that cocaine use disorder involves a redistribution of dopamine between vesicular and cytosolic stores (
Figure 3 is a graphical depiction of this hypothesis). Chronic cocaine exposure is associated with a reduction in vesicular monoamine transporter 2 (VMAT2) expression, which leads to less dopamine in the vesicular pool and more in the cytosolic pool. The reduction in VMAT2 has been shown in nonhuman primates who chronically self-administer cocaine (
27) and in human cocaine users (
28). Postmortem human studies also show a reduction of striatal VMAT2 in cocaine users (
29–
31).
Blunted VMAT2 expression in cocaine use disorder would explain the decrease in presynaptic dopamine release seen with PET (
1–
4) and could also account for the decrease in [
18F]DOPA accumulation seen in this population (
32), since this likely depends on the radiotracer concentrating in synaptic vesicles (
33). Reduced VMAT2 expression has also been shown to correlate with elevated NM formation in the midbrain (
12,
34). While cocaine use has been shown to be associated with altered expression of dopamine receptors and transporters (
1), likely including autoreceptors in the midbrain (
35), these changes would generally shift both NM accumulation and dopamine release in the same direction. VMAT2 alteration, on the other hand, stands out as a parsimonious explanation for the observed changes occurring in opposing directions. Taken together, these imaging studies suggest that cocaine use is associated with lower dopamine in the vesicular pool and a higher concentration in the cytosolic compartment. However, a study imaging VMAT2 and dopamine release in cocaine users combined with NM-MRI in the midbrain would be needed to confirm our hypothesis. If cocaine use indeed increases cytosolic dopamine, this may pose a risk to neurons, because oxidation of dopamine in this compartment forms reactive quinone species (
36); however, there is no clear evidence of enhanced dopamine cell death (
37) or Parkinson’s disease risk (
38) in cocaine users.
An alternative interpretation of our main finding is that NM elevation in cocaine users results from repeated episodic surges in dopamine that occurred over the participants’ lifetime, which may not be captured by PET. Since NM granules are removed only after cell death (
26), and thus serve as a long-term reporter of dopamine function, even a distant history of cocaine use (which may acutely lead to excess dopamine during cocaine consumption) could manifest as a persistent increase in the NM-MRI signal. Longitudinal studies would be needed to address this possibility.
As an initial test of the functional significance of our findings, we examined whether NM-MRI signal in cocaine-use voxels within the SN correlated with fMRI response to reward anticipation in the ventral striatum during the monetary incentive delay task, a robust probe of reward system function (
15,
19,
25). We failed to find a significant correlation. This is perhaps unsurprising since the abnormality that we observed in cocaine users was not clustered near the “limbic” SN or ventral tegmental area (dorsomedial regions of our overinclusive SN mask [
21]) that send the main projections to the ventral striatum. Rather, our topographical analysis showed that group differences predominated in the ventral (or “cognitive”) SN (
21), a subregion with prominent projections to the dorsal striatum thought to be involved in cognitive flexibility and other higher-order functions (
20,
21). While PET imaging studies of dopamine function in cocaine users have found consistent evidence of dopaminergic alterations in the dorsal striatum, they have also found pronounced alterations in the ventral striatum (
1). Intriguingly, our observation that cocaine users show an increase in NM-MRI signal in dorsal-striatum-projecting regions of the SN but not in ventral-striatum-projecting regions aligns with the previous observation of significant VMAT2 reductions in the dorsal but not the ventral striatum in this population (
28,
31). Whatever may underlie this anatomical pattern, it highlights that nigrostriatal circuits subserving cognitive functions may be important in cocaine use disorder and that future studies may be better positioned to determine the functional significance of NM-MRI signal change in this disorder by probing higher-order cognitive processes in addition to reward tasks.
The primary limitation of this study is the relatively small, entirely male, sample. However, we contend that this first report of NM-MRI in substance use disorders supports the promise of this method for measuring dopamine function in this population. The only previous NM-MRI study to investigate substance use was a preliminary evaluation of the size of the SN area in a small group of patients with psychotic illness. Psychotic patients with comorbid substance use exhibited a larger SN area than nonuser patients (
39). We are not aware of previous work investigating NM concentration in postmortem tissue in substance use disorders, and this would be an important future direction to provide convergent support for our findings. Based on the present results, we cannot speak to the generalizability of our NM-MRI findings in cocaine users to other substance use disorders. Further research is needed to address this question, especially in light of our findings showing a near-significant relationship between NM-MRI and tobacco use (which may well reach significance in a larger sample or in heavier tobacco users). Assuming that increased NM signal is due to down-regulation of VMAT2 (
27,
28), the reported NM-MRI phenotype may be specific to cocaine or other drugs affecting VMAT2 (perhaps including methamphetamine, although its relationship to VMAT2 is less clear [
1]). The absence of a significant correlation between NM-MRI signal and duration of cocaine use in our data is surprising. Given that NM accumulates over time, we anticipated that longer duration of use would exaggerate any abnormalities observed in cocaine users. The lack of a significant relationship could, however, be due to the limited range in duration of use in our sample, as our participants had all been using cocaine for many years. The NM-MRI signal does not reflect a single biological process but could be altered by changes in dopamine synthesis (
12), dopamine transfer to vesicles (
34), or dopamine cell death (
6). Such nonspecificity is common to imaging measures (
40,
41) and argues for the utility of multimodal studies in triangulating neurobiological mechanisms, as we have attempted to do by interpreting our findings in light of previous PET imaging reports. While interpretation of our NM-MRI results is simplified by the absence of enhanced dopamine cell death in cocaine users (
37), interpretation of NM-MRI results in disorders showing substantial cell death combined with altered NM accumulation may be more challenging.