In this issue of the
Journal, Zink et al. (
1) report remarkable new findings relating variance in the folate hydrolase 1 (
FOLH1) gene encoding for glutamate carboxypeptidase II (GCPII) to human intelligence. Although there have been many genetic insults that have been linked to severe intellectual disability, it is rare to find a genetic variant that is related to IQ within the normal range. The authors found that a missense mutation in
FOLH1 that leads to greater expression of GCPII in both healthy subjects and patients with schizophrenia is associated with reduced levels of its substrate,
N-acetylaspartylglutamate (NAAG), and with lower IQ. Consistent with lower intelligence, the authors found inefficient processing by the dorsolateral prefrontal cortex (DLPFC), a newly evolved cortical region that subserves working memory, abstract reasoning, and executive functioning.
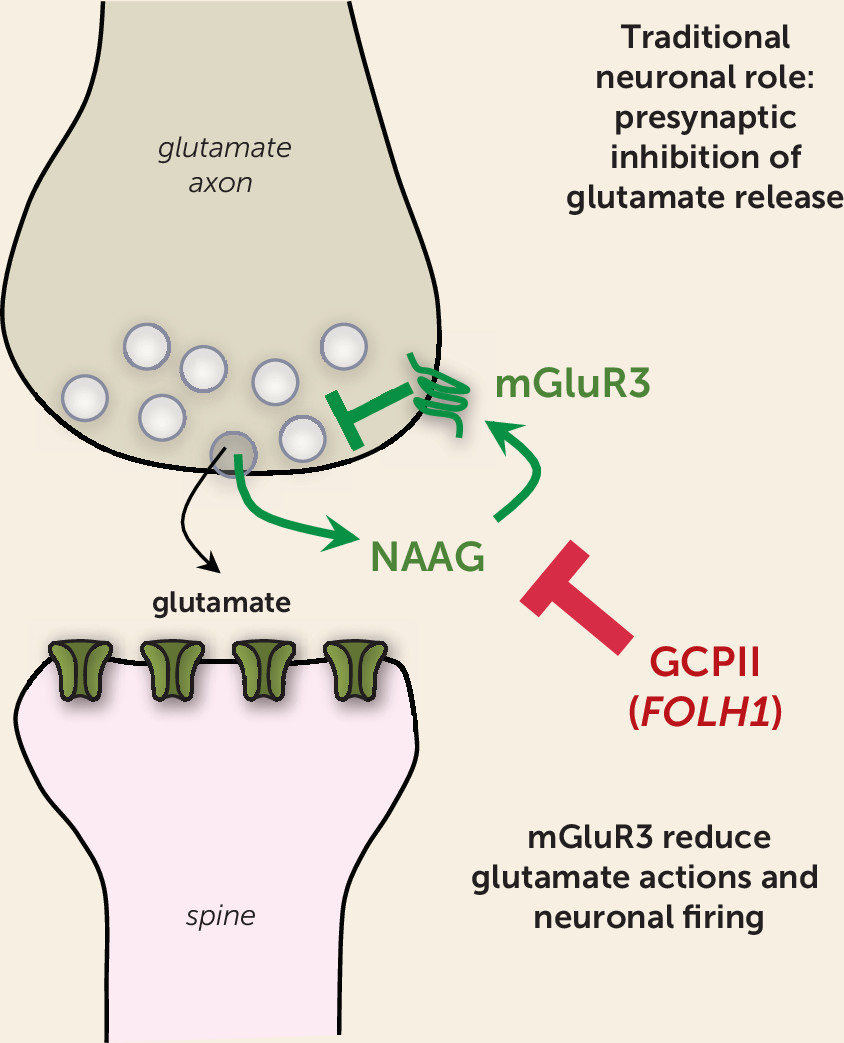
NAAG is a highly prevalent (yet underappreciated) neurotransmitter that is coreleased with glutamate (
Figure 1). Importantly, NAAG selectively stimulates metabotropic glutamate receptor type 3 (mGluR3, encoded by the
GRM3 gene). Genetic alterations in
GRM3 are consistently linked to increased risk of schizophrenia by genome-wide association studies (
2), further emphasizing the importance of mGluR3-GCPII signaling to human cognition and cognitive disorders. However, the links to higher cognition have been perplexing to many scientists, as mGluR3s are classically viewed as glial receptors.
mGluR3s are localized on astrocytes, where they enhance glutamate uptake through excitatory amino acid transporters (reviewed by Neale et al. [
3]). Studies in rodents show that they can also play a related role in classic neuronal circuits, where they often reside on presynaptic terminals and reduce glutamate release (
3) (
Figure 1). Thus, mGluR3s traditionally have been seen as providing negative feedback on glutamate signaling and can be protective against excitotoxicity in rodent models (
4). However, recent data indicate that their role in some neurons has changed and expanded with cortical evolution.
Unique Role of NAAG-mGluR3 Signaling in the Newly Evolved Cortical Circuits Subserving Higher Cognition
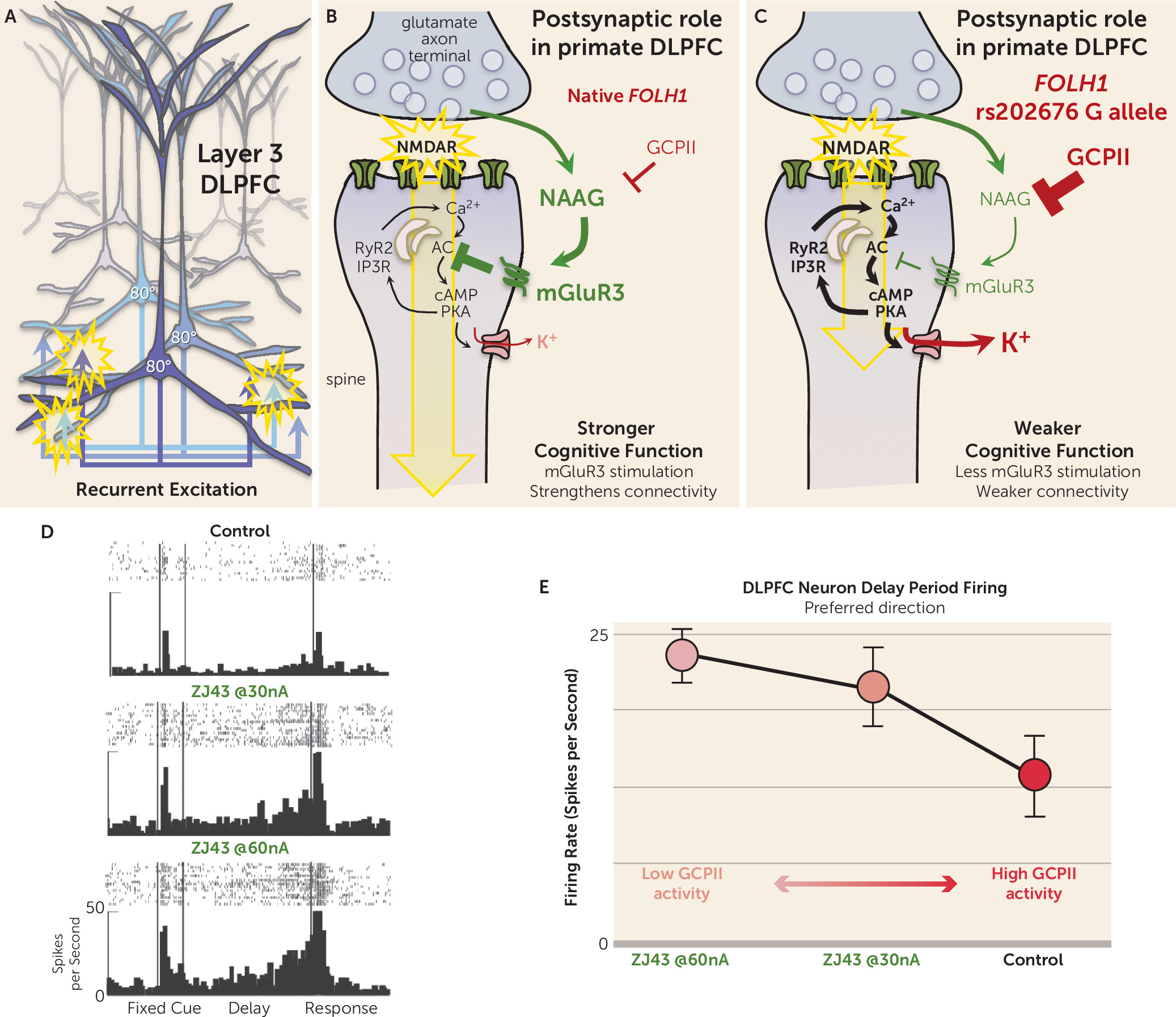
Studies of the rhesus monkey DLPFC have discovered that mGluR3 and GCPII have a novel role in higher cortical circuits in primates—strengthening the connectivity of layer 3 DLPFC circuits that mediate working memory—helping to explain their genetic links to cognition and cognitive disorders.
The prefrontal cortex (PFC) expanded greatly over brain evolution, especially the DLPFC and frontal pole regions that do not exist in rodents, and greatly increases in size across nonhumans to human primates. Research by Goldman-Rakic and Gonzalez-Burgos has shown that the primate DLPFC contains extensive recurrent excitatory connections in deep layer 3 (
Figure 2A), where pyramidal cells excite each other through N-methyl-
d-aspartate receptor (NMDAR) synapses (
Figure 2B) to keep information “in mind” (reviewed in [
5]). The information is refined by lateral inhibition from GABA interneurons to generate a precise representation. In this way, layer 3 microcircuits are able to maintain representations of information across a delay period without the need for sensory stimulation, the foundation of abstract thought. These NMDAR synapses are powerfully and uniquely modulated by the arousal systems (e.g., where exposure to an uncontrollable stressor can weaken synaptic connectivity by driving high levels of calcium-cyclic AMP-dependent protein kinase [cAMP-PKA] signaling), which in turn open potassium (K
+) channels near the synapse to decrease recurrent network excitation and reduce neuronal firing (
Figure 2B) (reviewed by Arnsten et al. [
5]). Recent data suggest that mGluR3 plays a major role in regulating these signaling events in primate DLPFC, inhibiting cAMP-PKA-K
+ signaling to enhance synaptic strength and persistent neuronal firing.
Immunoelectron microscopy (immunoEM) can reveal the subcellular location of proteins in brain. ImmunoEM of the rhesus monkey layer 3 DLPFC showed that mGluR3s are concentrated postsynaptically on spines (
Figure 2B), completely different from their classic location on presynaptic terminals in rodent circuits. mGluR3s are also localized on astrocytes in primate DLPFC, but the presynaptic receptors on glutamate axon terminals are exclusively mGluR2 rather than mGluR3 (
6). mGluR3s on spines can be found within the postsynaptic density but are mostly localized on the spine membrane near the calcium-containing spine apparatus, the extension of the smooth endoplasmic reticulum into the spine, which stores and releases calcium. As illustrated in
Figure 2B, cAMP-PKA signaling drives calcium release from the spine apparatus, which in turn stimulates more cAMP production, thus creating a rapid rise in calcium-cAMP-PKA signaling. PKA signaling can also increase calcium flux through NMDAR and through voltage-gated calcium channels, such as Cav
1.2, magnifying cytosolic calcium (reviewed by Arnsten et al. [
5]). As mentioned above, high levels of calcium-cAMP-PKA signaling open nearby K
+ channels to weaken connectivity and reduce neuronal firing. Conversely, inhibition of cAMP signaling closes K
+ channels, strengthens connectivity, and enhances the persistent neuronal firing needed for working memory (
5,
7). These beneficial actions have been seen with NAAG-mGluR3 signaling in primate DLPFC, where iontophoretic application of either NAAG or a GCPII inhibitor (
Figure 2D and E) directly onto DLPFC neurons greatly enhances working memory-related neuronal firing by inhibiting cAMP-PKA-K
+ channel signaling (
6). This can be seen as a dose-response, where increasing amounts of GCPII activity lead to decreasing levels of DLPFC neuronal firing. Thus, NAAG-mGluR3 signaling strengthens the connectivity of higher cortical glutamatergic circuits and increases DLPFC neuronal firing in primates, opposite to the reduction in glutamate release classically associated with mGluR3 presynaptic actions in rodents.
The new study by Zink et al. highlights the consequences to cognition when a genetic variation increases the expression of GCPII and reduces NAAG levels in brain. Consistent with a reduction in beneficial NAAG-mGluR3 actions, the authors found a positive correlation between NAAG levels measured by magnetic resonance spectroscopy and cognitive performance. The study also found that carriers of the missense
FOLH1 variant had inefficient activation of DLPFC during working memory and lower IQ scores. Coupled with the findings in monkeys, the data suggest that the human subjects with the
FOLH1 variant would have less NAAG stimulation of mGluR3, greater opening of K
+ channels, and weaker DLPFC network connectivity needed for working memory and abstract reasoning (
Figure 2C) and thus would have to recruit greater amounts of DLPFC to perform a cognitive task (i.e., inefficient action of the DLPFC as seen in the Zink et al. study). Because GCPII expression is also increased by inflammation, we can speculate that similar insults to cortical connectivity and cognitive function may occur during an infection, perhaps helping to explain why we often have “brain fog” during illness.
Relevance to Cognitive Disorders
The results from human and monkey studies suggest that either genetic or environmental insults that increase GCPII and/or reduce NAAG-mGluR3 signaling can weaken higher cortical connections and increase risk of cognitive disorders. For example, there is an increased risk of schizophrenia with mutations to
GRM3 (
2), or with perinatal inflammation, which in animal studies, increases GCPII expression in the brains of offspring (
8). Other genetic insults that increase risk of schizophrenia may create a similar phenotype of increased cAMP-calcium signaling in the DLPFC (e.g., duplication of
VIPR2 [
VPAC2] or loss of function translocation in
DISC1 anchoring of phosphodiesterase 4 [PDE4] has been shown to increase cAMP signaling, while alterations in
CACNA1C [Cav1.2] increase calcium flux [reviewed by Arnsten et al. [
5]). While these signaling events are needed for normal function, excessive cAMP-calcium signaling in layer 3 DLPFC circuits is detrimental, opening large numbers of K
+ channels to reduce firing and likely contributing to dendritic atrophy when sustained by chronic conditions (
5).
The increase in GCPII expression with inflammation may also contribute to cognitive deficits in aging and Alzheimer’s disease (
9). Advancing age is associated with loss of PDE4 (
10) and mGluR3 (
11) regulation of cAMP-calcium signaling in the aged PFC. Studies of aging rhesus monkeys have shown that this dysregulated cAMP-PKA-K
+ signaling in the DLPFC contributes to age-related loss of persistent neuronal firing needed for working memory (
12) and to phosphorylation of tau, which leads to neurofibrillary tangles (
10). Elevated GCPII signaling also appears to contribute to cognitive deficits in multiple sclerosis, where hippocampal NAAG levels correlate with cognitive abilities (
13). It is possible that elevated GCPII due to infectious conditions, such as COVID-19, may also contribute to the delirium and cognitive deficits that can accompany this disease.
Relevance to Potential Treatments
GCPII inhibitors are currently under development for the treatment of inflammatory and cognitive disorders (
14). For example, they have shown success in animal models of neuropathic pain, Alzheimer’s disease, and multiple sclerosis (
14–
16), and studies of aging monkeys are in progress. Assays of GCPII inhibitors in human and animal studies to date have shown that these compounds appear to have excellent side-effect profiles, encouraging their use and potential utility for disease prevention. However, current compounds have limited brain penetrance, and this will need to be overcome to be of practical benefit in cognitive disorders.
In closing, the findings by Zink et al., coupled with the links between GRM3 and schizophrenia, serve as genetic “flashing lights,” alerting us to the importance of a previously underappreciated signaling pathway important to human cognition. These genetic associations are further illuminated by the recent data from macaque DLPFC showing that NAAG-mGluR3 signaling strengthens network connectivity in the circuits that generate the mental representations underlying working memory and abstract reasoning. Taken together, they provide a rational story, where the role of GCPII-NAAG-mGluR3 signaling expands with cortical evolution to regulate the circuits underlying human intelligence.