In 1976, I (J.T.C.) finished my psychiatric residency, received my first National Institutes of Health grant, opened my laboratory, and took responsibility for the Schizophrenia Outpatient Clinic at the Johns Hopkins Phipps Clinic, my first academic clinical responsibility. That year was a pivotal point in the evolution of scientific psychiatry, as the dopamine hypothesis of schizophrenia (
1) was proposed, the first report of synaptic neurochemical pathology of schizophrenia appeared (
2), and Eve Johnstone and colleagues first demonstrated cortical atrophy in schizophrenia using computerized axial tomography (
3). In this overview article, we review the seminal advances that have fundamentally transformed how we understand the etiology of schizophrenia and related serious mental disorders over the past 50 years, which will thus transform psychiatric diagnosis and treatment.
The 1970s witnessed the transition from the hegemony of psychoanalysis, with virtually every chairperson of an academic department of psychiatry in the United States being a psychoanalyst, to the rise of “biologic” psychiatry. However, biologic psychiatry might be more accurately designated “psychopharmacologic” psychiatry, as serendipitously discovered drugs found to be relatively effective treatments for serious mental disorders, including schizophrenia (neuroleptics), bipolar disorder (lithium), depression (tricyclics), and anxiety disorders (benzodiazepines), essentially guided research with an unspoken assumption that the mechanism of action of these drugs was closely linked to the pathophysiology of the disorders. Notably, the decade was ushered in by the award of the Nobel Prize to Julius Axelrod (my postdoctoral mentor) for the discovery that tricyclic antidepressants inhibit the uptake of norepinephrine into noradrenergic synaptic terminals (
4)—and thus norepinephrine became the “depression” neurotransmitter (
5).
For example, chlorpromazine, the prototype antipsychotic, was initially developed as an adjunct to anesthesia. Noting its calming effects on surgical patients, the French psychiatrists Delay and Deniker (
6) carried out a clinical trial of chlorpromazine in patients with schizophrenia and described striking improvements in thinking and behavior unrelated to sedation. The pharmaceutical industry undertook studies in experimental animals to identify the behavioral “signature” for the antipsychotic action of chlorpromazine to screen for more potent drugs with different side effect profiles. Over the next decade, several more potent antipsychotics, with different chemical structures, such as haloperidol, molindone, and fluphenazine, were introduced (
7).
In 1963, the Swedish pharmacologist Arvid Carlsson discovered that antipsychotic drugs interfere with dopamine signaling in the rat basal ganglia and proposed that their mechanism of therapeutic action is through blocking the dopamine receptor (
8). In 1976, Solomon Snyder built on this observation to show that there is highly significant correlation between antipsychotics’ efficacy in the clinic and their affinity for the brain’s dopamine D
2 receptor (
9). This observation was the basis for the “dopamine hypothesis of schizophrenia,” which proposed that dopaminergic neuronal dysfunction is the cause of schizophrenia (
1). The hypothesis gave me enormous hope that the rational use of antipsychotics, including determination of their blood levels by radioreceptor assays that measure the drug’s occupancy of the dopamine D
2 receptor in a test tube, would optimize treatment of patients with schizophrenia (
10), and monitoring the drugs’ anticholinergic effects could limit side effects (
11).
After several years of perfecting this strategy, there were some minor victories as the patients tended to have reduced problems with unresponsive psychosis (nonadherence was easily detected), reduced extrapyramidal side effects, and less cognitive impairment (
12). Nevertheless, no patients were discharged as “well,” and most remained severely disabled, estranged from their families, unemployed, and isolated. These observations corresponded to the findings that cognitive impairments and negative symptoms such as anhedonia, amotivation, and asociality were core symptoms of schizophrenia that did not respond to the antipsychotic drugs (
13,
14). Furthermore, since the introduction of antipsychotic drugs, recovery from schizophrenia has changed little except for reduced psychotic symptoms (
15).
One bright spot was the resurrection by academic investigators of clozapine (
16), an off-patent drug, which was found to be effective with some patients who were unresponsive to other antipsychotics and which reduces the risk of suicide (
17) and substance abuse (
18). The greater efficacy of clozapine, which interacts with multiple neurotransmitter systems, suggested that neurotransmitter systems beyond the dopamine D
2 receptor might have an impact on negative symptoms and cognitive impairments (
19). Clozapine also ushered in the “second generation” of antipsychotic drugs that maintained the dopamine D
2 receptor antagonism but dialed in serotonin receptor interactions that attenuated extrapyramidal side effects (
20).
If schizophrenia is a brain disorder, then pathology should be evident in the brain. Studies using vital stains developed in the late 19th century to visualize cell integrity in postmortem brain tissue were instrumental in identifying sites of pathology and neuronal loss in the emerging field of neurology (
21). However, these methods provided little insight into the cause of symptoms in psychiatric disorders (
22), leading the neurologist Fred Plum to opine that “schizophrenia was the graveyard for neuropathologists” (
23). However, the relationship between postmortem neuropathology and symptoms remained unclear even for some neurologic conditions like Parkinson’s disease. In 1960, Oleh Hornykiewicz carried out a survey of the levels of dopamine and norepinephrine in the postmortem human brains of three patients who died of Parkinson’s disease, and found a profound loss of dopamine in the caudate and putamen (
24). These results indicated that there could be important “neurochemical” pathology in the absence of obvious cellular pathology.
Following Hornykiewicz’s lead, Edward Bird and colleagues at the University of Cambridge reported in 1977 the results of the first neurochemical study on postmortem brains from 41 individuals with psychosis (
2). They found elevations of dopamine in limbic regions, but they also found reductions in the activity of glutamic acid decarboxylase, the biosynthetic enzyme for γ-aminobutyric acid (GABA), in the hippocampus, amygdala, and nucleus accumbens, and reduced activity of choline acetyltransferase, the biosynthetic enzyme for acetylcholine, in the nucleus accumbens. While the elevations in dopamine were consistent with the dopamine hypothesis, the reductions in GABAergic and cholinergic markers indicated a broader neural pathology than just disruption of dopaminergic neuronal function.
With the development of multiple methods for measuring gene expression and the expansion of brain banks with standardized collection techniques, the past 30 years of research have yielded a body of consistent results with concurrence across modes of analysis, including quantitative neurochemistry, in situ hybridization, immunocytochemistry, and DNA chip array. For example, several groups have developed consistent evidence of a down-regulation of presynaptic markers for the fast-firing parvalbumin (PV+)-expressing GABAergic interneurons and up-regulation of their postsynaptic GABA
A receptors (
25–
27), indicative of reduced GABAergic recurrent inhibition of cortical pyramidal neurons.
In light of the evidence from postmortem Golgi stain studies showing the loss of dendritic spines, which are excitatory glutamatergic synapses, studies increasingly addressed components of the glutamatergic synapse in corticolimbic regions of the schizophrenic brain (
28). Meador-Woodruff and colleagues reviewed the postmortem findings on the alterations in glutamate receptor trafficking,
N-methyl-
d-aspartic acid (NMDA) receptor subunit levels, and postsynaptic density-associated proteins of the glutamatergic synapse in schizophrenia (
29,
30). Catts et al. (
31) carried out a meta-analysis of nine studies that measured the mRNA and/or protein for the NR1 channel component of the NMDA receptor and found highly significant reductions in the prefrontal cortex. Although a wide array of abnormalities in markers for different neurotransmitter systems, including dopaminergic, serotonergic, and cholinergic neurons, have also been reported, it has been difficult to distinguish whether they might be caused by the disorder itself, be a result of years of exposure to psychotropic medications, be the consequence of substance abuse (which affects the majority of patients [
32,
33]), or result from comorbid medical conditions, such as diabetes mellitus (
34).
With the development of brain imaging methods, including CT, PET, and MRI, it became possible to view the structure, chemical composition, and function of the brain in the living individual. An initial finding of great importance in schizophrenia research was the CT study of Johnstone and colleagues in 1976 showing ventricular enlargement in a cohort of patients with chronic schizophrenia as compared with matched control subjects (
3). This result confirmed clinical reports using pneumoencephalographic X-ray methods and early postmortem studies (
35). A meta-analysis of 39 studies using structural MRI to quantify ventricular volume carried out 15 years later confirmed the ventricular enlargement, commenting that it was “an indisputable characteristic of schizophrenia” (
36).
Since brain volume is constrained by the cranium, an increase in ventricular volume would imply a loss of brain mass. Many studies using quantitative structural MRI describe volume reductions in cortical subregions, including the hippocampus (
37), temporal cortex (
38), and prefrontal cortex (
39), leading the field to focus on these regions because of their roles in memory, emotional control, and executive functions, which are disrupted in schizophrenia. However, Kuperberg et al. (
40), utilizing high-resolution structural MRI, developed compelling evidence that cortical thinning is not circumscribed but rather involves the entire cerebral cortex. The fact that there is no difference in skull size between adult control subjects and those with schizophrenia suggests that much of the atrophy occurs after substantial brain maturation has occurred. Studies done early in the course of psychosis as well as studies in ultra-high-risk youths who transition to psychosis reveal detectable cortical atrophy (
41); but longitudinal studies indicate that the atrophy progresses for at least a decade after the onset of psychosis (
42).
These two findings—that cortical atrophy, including gray and white matter, is present at psychosis onset and that atrophy progresses for at least a decade afterward—raised the question of whether schizophrenia is a neurodegenerative disorder like Huntington’s disease. Rigorous stereologic cell counting techniques and other methods for assessing neuronal loss in cortex and hippocampus in schizophrenia have not revealed significant neuronal loss, at least not commensurate with the degree of cortical volume reduction (
43). Returning to the findings from postmortem studies, generally involving patients decades after symptom onset, investigators report increased cell-packing density, reduced neuronal somal cell size, reduced myelinated tracts (
44), and, in Golgi studies, reduced dendritic length and reduced synaptic spine density (
28,
45,
46). The cortical atrophy affects primarily the space between neurons, the neuropil, which consists primarily of axons, dendrites, and synapses (
47,
48). By taking into account the loss of dendritic length and the loss of synaptic spines, the absolute loss of cortical glutamatergic synapses is estimated to be in the range of 30%, much greater than the degree of cortical atrophy (
45).
From the beginning, Kraepelin and Bleuler argued that genetics played an important role in the cause of schizophrenia (
49). A series of family and twin studies initiated nearly 100 years ago, as reviewed in a landmark pair of articles by Seymour Kety published in
Science in 1959, provided compelling evidence of a high degree of heritability of schizophrenia (
50,
51). Approximately 60% of identical twins and 15% of fraternal twins and first-degree relatives exhibit concordance for schizophrenia, consistent with a non-Mendelian mechanism of inheritance or complex genetics (
52). At the turn of this millennium, with the completion of the sequencing of the human genome, attention had turned to identifying “risk genes” for schizophrenia. Risk genes are typically noncoding variants that significantly increase the probability of developing schizophrenia and interact with other risk genes (epistasis) and the environment to produce the phenotype (
53).
Not surprisingly, early studies focused on genes encoding proteins implicated by the dopamine hypothesis known as “candidate genes.” For example, using the case-control method (patients compared with control subjects), several studies involving at most 100 subjects implicated the dopamine D
2 receptor gene and related dopaminergic core genes as risk genes for schizophrenia (
54). Meta-analysis of the results of these studies to boost statistical power suggested compelling significance. But this strategy proved misleading, as selection of candidate genes was biased, the studies were grossly underpowered (leading to type I statistical errors), and publication bias, where negative findings were either not submitted to journals or were rejected by them, inflated the impact of positive results (
54).
As psychiatric genetics matured, the approach shifted to genome-wide association studies (GWASs), which enforced a much more rigorous statistical threshold of 5×10
−8 to correct for the multiple comparisons inherent to screening the entire genome unbiased by etiological theories (
55). The distribution of single-nucleotide polymorphisms (SNPs) was mapped on the chromosomes, revealing a frequency of 1 in 1,000 base pairs and a low risk of mutation. SNPs became the favored marker for doing association studies. To reach this high statistical threshold, it is necessary to study many thousands of patients and control subjects. This challenge required a new approach to research to accumulate such large numbers. Since no single clinic could provide more than a few hundred clinically well characterized patients, a consortium of dozens of research clinics from several countries had to pool their subjects to achieve the required number (the Schizophrenia Working Group of the Psychiatric Genomics Consortium).
The first major GWAS on schizophrenia, published in 2014 and involving genomes from 38,000 subjects with schizophrenia and 110,000 control subjects, indicated that 108 sites on the genome achieved the 5×10
−8 level of significance (
55). Most recently, the cohort has been expanded to nearly 70,000 schizophrenia patients and 236,000 control subjects, which yielded 270 loci associated with significant risk (
56). Fine-mapping identified 130 genes (approximately 0.5% of the human genome) that confer risk for schizophrenia. A clear pattern has emerged. Most of the risk SNPs do not localize to coding regions of the genome but presumably act by altering gene expression. A few highly penetrant sites are copy number variants (CNVs) that result from deletion or replication of kilobases of DNA and include multiple genes. Kirov et al. (
57) found that CNVs associated with schizophrenia are significantly enriched with genes encoding proteins localized to the glutamatergic synapse and its postsynaptic density.
The bulk of the identified risk genes are implicated in neuronal differentiation, organization, and transmission (
56). Almost 30% of the 130 risk genes encode proteins that cluster around the pre- and postsynaptic components of the glutamatergic synapse and would affect NMDA receptor transmission.
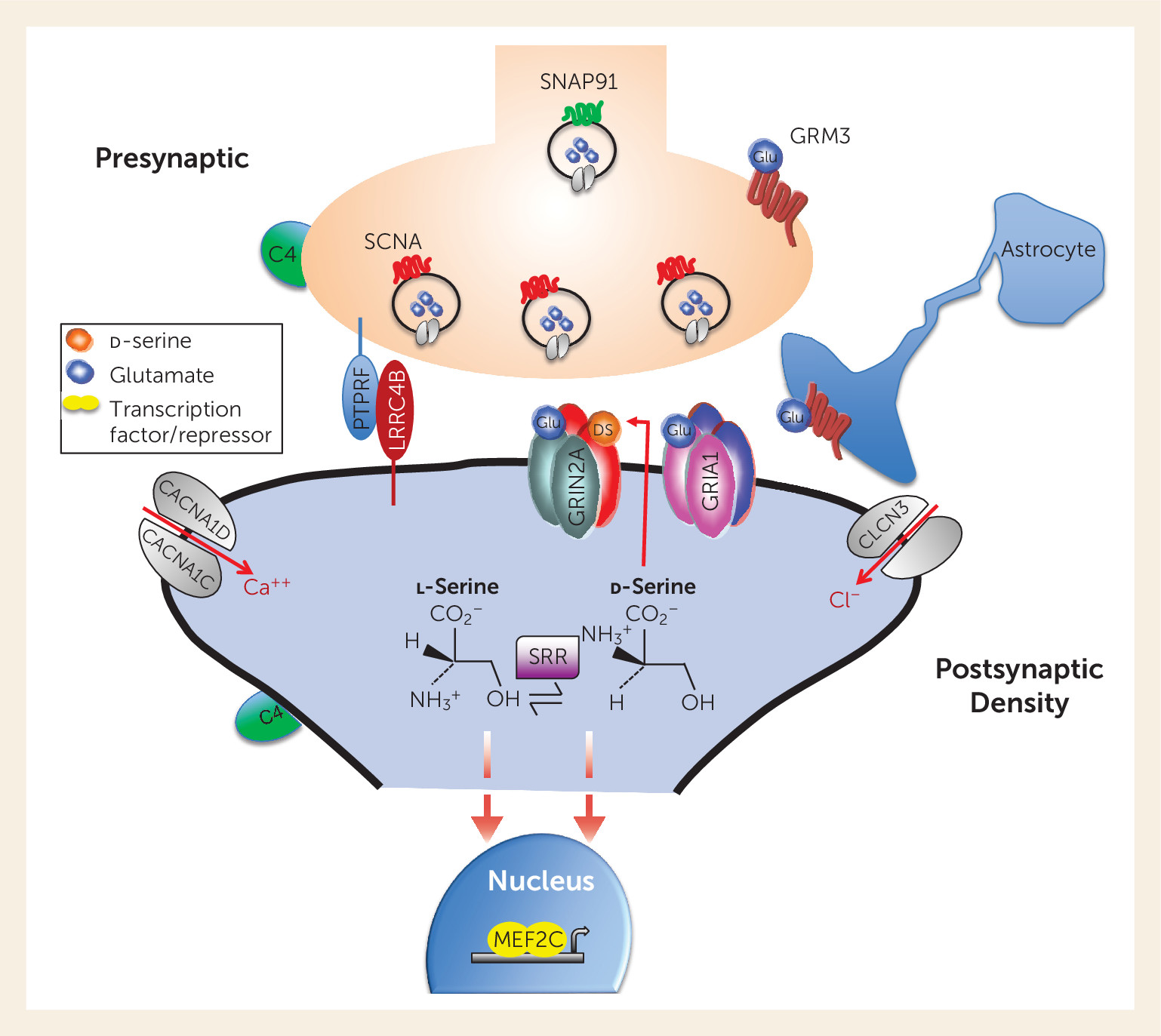
Figure 1 illustrates the localization of several of these risk genes at the glutamatergic terminal, its postsynaptic spine, and downstream effectors. Several more are associated with the GABAergic neurons. Ironically, only one gene related to the dopamine hypothesis, the one encoding the D
2 receptor, has survived the rigorous GWAS analysis, consistent with the evidence that dopamine dysregulation is likely a mediator of psychosis (
58) but is not explanatory for the full schizophrenia syndrome. Recently, by meta-analyzing whole exomes (the coding regions of the genome), Singh et al. (
59) reported 10 coding mutations that conferred substantial risk for schizophrenia, including genes encoding the subunits of the NMDA and AMPA (α-amino-3-hydroxy-5-methylisoxazole-4-propionate) receptors, further substantiating the centrality of the glutamatergic synapse in the etiology of schizophrenia.
The concordance rate of approximately 60% for schizophrenia among monozygotic twins demonstrates a significant influence of nongenetic or environmental factors in mediating the phenotypic outcome of genetic risk variants (
52). One of us (W.B.R.) has focused his laboratory on the impact of the environment on the genome and gene expression, known as epigenetics, with an emphasis on schizophrenia. Epigenetic mechanisms—chemical and structural modifications to chromatin that influence gene expression without alterations in the DNA base sequence—have emerged throughout medicine as ubiquitous mediators of the molecular interaction between the static genome and dynamic environmental events (
60). For example, adults who experienced abuse as children have altered epigenetic states and decreased expression of the glucocorticoid receptor gene
NR3C1 in the hippocampus as compared with control subjects without a history of childhood abuse (
61,
62). Epigenetic mechanisms are especially important in the complex pathophysiology of psychiatric disorders, including schizophrenia, where they mediate the emergence of molecular pathologies in specific populations of cells in specific brain regions stemming from nonspecific genetic and environmental risk factors. Fetal viral infections, famine during pregnancy, and complicated birth (
63) as well as childhood abuse and neglect (
64) are established environmental risk factors for schizophrenia.
DNA methylation occurs in mammalian genomes on cytosine residues within CpG dinucleotides and has variable effects depending on the genomic context of the methylated DNA sequence. For instance, promoter methylation is associated with decreased gene expression, while the gene body of active genes typically shows higher methylation than that of silenced genes (
65), and DNA methylation is known to have an impact on RNA splicing through multiple mechanisms (
66). Case-control comparison of DNA methylation levels in the prefrontal cortex has identified >2,000 differentially methylated loci across the genome in schizophrenia relative to control subjects, and these are associated with nearly 60% of known schizophrenia GWAS loci (
67). The greatest change in genome-wide DNA methylation patterns occurs at the fetal-neonatal transition, involving many of the sites that are linked to schizophrenia GWAS loci, consistent with the role of neurodevelopmental and perinatal insults in schizophrenia (
67,
68). DNA methylation is altered in specific cell populations and circuit locations within the brain in schizophrenia, with selective overactivity of the DNA methylation machinery in layer I of the prefrontal cortex, and subfield-specific dysregulation of DNA methylation at genes relevant to GABAergic interneurons in the hippocampus (
69).
In addition to covalent modifications of chromatin through methylation and other mechanisms, epigenetics also regulates gene function through modifying the large-scale three-dimensional conformation of the chromosomes. Through regulated chromatin looping, genetic loci that are physically distant within the two-dimensional DNA sequence can be brought into close physical proximity, permitting interaction between factors recruited to an enhancer and its target promoters. The three-dimensional architecture of the genome is again highly cell type-specific and dynamic across development, and developmental changes at schizophrenia-implicated genomic loci occur disproportionately in neurons (
70). This as-yet-underexplored mechanism is argued to increase the “three-dimensional genome risk space” for schizophrenia by facilitating interaction between noncoding schizophrenia risk loci and genes more physically distant than are typically considered putative targets (
68). These early findings in the new area of epigenetics give a glimpse of how incredibly complex the intersection between genomics and epigenomics will be.
Since its inception, J.T.C.’s laboratory has focused on the role of glutamate in neuropsychiatric disorders (
71). Twenty-five years ago, we published a postmortem analysis that revealed alterations in the neurochemistry of the cortex and hippocampus in schizophrenia consistent with impaired NMDA receptor–mediated neurotransmission (
72). A year earlier, John Krystal’s laboratory published the results of a study with healthy volunteers showing that infusion of low doses of ketamine, a dissociative anesthetic that is an NMDA receptor antagonist, caused negative symptoms (emotional blunting), subtle cognitive impairments similar to those of schizophrenia and without delirium, and misperceptions consistent with early stages of hallucinations, thus replicating the complete phenotype of schizophrenia (
73). Subsequent studies with ketamine infusions extended these similarities to hypofrontality in performing a cognitive task, increased dopamine release with amphetamine challenge, and EEG abnormalities similar to schizophrenia (
74–
76). The confluence of the clinical and postmortem findings prompted us to focus on how NMDA receptor hypofunction might affect the brain, using transgenic mice (
77).
The NMDA receptor is sui generis, as it is the only receptor that requires three simultaneous events for the receptor to transduce a signal: 1) the neuronal membrane must be depolarized; 2) the co-agonist,
d-serine, or the weaker ligand, glycine, must be bound to its recognition site on the NR1 subunit; and 3) the neurotransmitter glutamate must be bound to its recognition site on the receptor (
78). These functional constraints, unique to the NMDA receptor, indicate how important is the fidelity of NMDA receptor function. NMDA receptor activation has neurotrophic effects, is responsible for use-dependent synaptic plasticity, known as long-term potentiation, and stimulates the formation and maintenance of glutamatergic synapses (
79,
80). Thus, the NMDA receptor is
the driver of neuroplasticity in the brain. To impair NMDA receptor function without directly eliminating the receptor, we chose to knock out the expression of serine racemase (SR), the enzyme that synthesizes
d-serine (
81,
82), using standard transgenic methods (
83). In part, our decision was driven by the reports of reduced brain SR (
84) and
d-serine in plasma and CSF in subjects with schizophrenia (
85–
88).
The mice, which are homozygous for SR−/−, have a 90% reduction in cortical
d-serine and a 70% reduction in NMDA-receptor-dependent long-term potentiation (
83,
89). They exhibit mild hyperactivity, a rodent surrogate for psychosis (
83). They have significant memory impairments that are associated with frontal cortical (
90), amygdala (
91), and hippocampal dysfunction (
89); significant cortical atrophy of 5% accompanied by a 20% increase in ventricular volume on quantitative MRI (
92); a down-regulation of PV expression in cortical GABAergic neurons; loss of the perineural nets surrounding the GABAergic neurons; and elevated oxidative stress (
93). Golgi staining reveals significant reduction of dendritic complexity and synaptic spine density in the frontal cortex (
90), sensory cortex (
94), and hippocampus (
89), which in aggregate indicate a loss of approximately 30% of cortical glutamatergic synapses (
94,
95). As in schizophrenia, SR−/− mice show significant sexual dimorphism, with females significantly less affected than males (
82).
Table 1 summarizes the remarkable similarities to the phenotypic characteristics of schizophrenia. Notably, treatment of adult SR−/− mice for 3 weeks with
d-serine to normalize its brain levels results in restored long-term potentiation, reversal of memory deficits, correction of cortical neurochemical abnormalities, and partial restoration of dendritic spines (
89,
96).
An alternative strategy for reducing NMDA receptor function is to create mice in which the expression of the NR1 subunit, which forms the receptor’s channel, is severely reduced. One method simply created mice heterozygous for an NR1 knockout, NR1+/− (
97,
98). In another, the expression of NR1 was reduced to approximately 10% of normal by inserting a neomycin resistance gene in a noncoding region of the gene encoding NR1 (NR1
neo/neo mice) (
98). These mice exhibit impaired spatial cognition, stereotypical behaviors, and reduced social interactions that can be reversed by treatment with clozapine (
99). Furthermore, they have behavioral and neurophysiologic abnormalities, including impaired prepulse inhibition (a warning tone reduces startle reflex to a subsequent loud tone, a test for assessing gating of sensory stimuli), increase in the latency to the N1 auditory response as measured by EEG, and reduced gamma synchrony, observed in schizophrenia (
100). Notably, knocking out the NMDA NR1 subunit only in cortical PV
+-GABAergic interneurons reproduces many of the neurocognitive, behavioral, and electrophysiologic abnormalities observed with the universal NR1 subunit knockdown model, indicating the importance of the PV
+-GABAergic neurons, especially with regard to the cortical electrophysiologic dysregulation associated with schizophrenia (
101). However, this highly focused elimination of NMDA receptor function in GABAergic neurons alone does not account for the loss of dendritic spines on pyramidal neurons and cortical atrophy occurring in schizophrenia.
Recently, a mother and adult son who both developed a psychotic disorder were found to have a CNV spanning 9p24.1, which represented a de novo mutation in the mother (
102). The CNV resulted in triplication of
GLDC, which encodes glycine oxidase, the enzyme that degrades glycine. Glycine is both a co-agonist at the
d-serine site on NR1 and a precursor to
d-serine. Thus, this mutation would result in reduced occupancy of the
d-serine site on NR1 and reduced NMDA receptor function. Mice were generated that were transgenic for the homologous CNV region and had four copies of
Gldc. Astrocytes cultured from the mutant mice showed a greater than twofold increase in the expression of glycine oxidase at 7 days of culture and significant reductions in glycine and
l-serine (
103). In a double-blind placebo crossover and subsequent open-label study in the mother and son, glycine (which doubled plasma
l-serine levels) and
d-cycloserine reduced symptoms as assessed with the total Brief Psychiatric Rating Scale (BPRS) score (
102), indicating that NMDA receptor hypofunction contributes substantially to the BPRS phenotype in schizophrenia.
Convergent pathology by strikingly different mechanisms reinforces the hypothesis that the loss of cortico-limbic glutamatergic synapses is the fundamental pathogenic feature in schizophrenia. “Pruning” by the action of complement C3 and C4 plays an important role in reducing superfluous synapses during cortical development, especially during adolescence (
104). The most robust association signal in the 2014 GWAS (
55) is located in a portion of chromosome 6 known as the major histocompatibility complex, which is enriched with genes related to immunity and has long been known to be associated with the risk for schizophrenia (
105). Fine mapping indicated that the responsible gene encoded complement C4 with the allelic variant linked to schizophrenia, causing higher expression (
106). C4 is expressed by neurons, localized to axons, dendrites, and synapses. C4 activates complement C3, allowing C3 to covalently bind to synaptic spines and promote their engulfment by phagocytic microglia (
106). Thus, in the case of individuals with schizophrenia, the C4 risk variant may result in overexuberant synaptic spine pruning, causing cortical atrophy and cognitive impairments.
These studies highlight the fact that the effects of mutations in neuronally expressed genes are not limited to the affected neuron but radiate throughout the nervous system because neurons exist in synaptic networks. The reduction in NMDA receptor function disrupts a series of elements in a circuit starting with cortical pyramidal neurons, the main output neurons in the cortex, impairing synapse formation, plasticity, and maintenance (
107). The hypofunction of NMDA receptors on the PV
+-GABAergic neurons blocks their mechanism of monitoring excitatory input, resulting in reduced GABAergic inhibitory feedback to the pyramidal neurons and increased pyramidal firing (
108). As shown above, these cortical disturbances account for cognitive impairments and negative symptoms. The disinhibited excitatory output from the limbic cortex drives subcortical dopamine release (
109,
110), which is manifested by hyperactivity in experimental animals and psychosis in humans (
111). Viewed from this perspective, schizophrenia is clearly a disorder of primary cortical pathology, with psychosis being a downstream consequence. Thus, antipsychotic drugs do not address the primary pathology of schizophrenia, as evidenced by persistence of cognitive impairments and negative symptoms in patients receiving optimal doses of antipsychotics.
Soon after the initial proposal of the hypothesis of NMDA receptor hypofunction in schizophrenia, double-blind placebo-controlled clinical trials of agents that enhance NMDA receptor function through the
d-serine site were undertaken by our group and others. Both
d-cycloserine (
102,
112,
113), a partial agonist, and glycine (
102,
114) and
d-serine (
115), full agonists, were shown in several small studies to significantly reduce negative symptoms and enhance cognition in patients with chronic schizophrenia who were receiving stable doses of antipsychotic drugs. A meta-analysis of the results of 26 blinded clinical trials of these agents acting at the NMDA receptor
d-serine site carried out over the following decade revealed highly significant reductions in overall psychopathology, with an effect size of 0.40, including negative symptoms, cognition, and even positive symptoms (
116). The effect on positive symptoms is noteworthy because the patients were already receiving optimal doses of antipsychotic drugs, which again indicates that positive symptoms are related to NMDA receptor hypofunction. Enhancing NMDA receptor function in schizophrenia reverses a cognitive deficit associated with underactivation of the superior temporal gyrus (
117) and with EEG deficits seen in mismatch negativity paradigms (
118).
While these results appear promising, they are more supportive of the proof of principle of NMDA receptor hypofunction than of a method of treatment because of several limitations. Chronic treatment with
d-cycloserine results in desensitization and loss of efficacy (
119). The
d-amino acids are weak, requiring substantial doses that are not palatable.
d-serine has the potential to cause renal tubular necrosis and causes NMDA receptor internalization with constant exposure. These limitations have prompted the search for drugs that could pharmacologically enhance NMDA receptor function or its downstream mediators. Since glycine is an agonist at the
d-serine site, an inhibitor of the glycine transporter (GlyT1) should thus enhance NMDA receptor function (
120). However, the one inhibitor that has progressed through phase 3 clinical trials failed (
121). An mGluR 2/3 receptor agonist to inhibit excess subcortical glutamate release, tested in a large clinical trial, also failed (
122). Both studies were designed to include any subject diagnosed with schizophrenia. Given that glutamate-related risk genes are likely to affect only a subgroup of individuals with schizophrenia, success might be improved by targeting those whose genome is enriched for these risk genes. This is now being accomplished by determining polygenic risk scores, that is, by focusing on a relative enrichment of risk genes in individual subjects included in a clinical trial of a “pro-glutamatergic drug” (
123).
In closing, over the past 50 years, concepts of the etiology of schizophrenia have evolved from purely psychologic, through the belief that antipsychotic drugs should reveal the cause, to emphasizing that rigorous, agnostic statistical analyses of genetic and epigenetic data would provide a more accurate account. As summarized here, current findings provide a very different conceptualization of schizophrenia than the one broadly held in 1976.
First, the identification of at least 130 genes that confer significant risk for schizophrenia indicate that the DSM-5 diagnosis encompasses a genetically diverse range of disorders, with individuals varying in their load of particular risk genes and environmental insults. Second, the primary neuropathology is a substantial loss of glutamatergic synapses in the cortex that is associated with a progressive cortical atrophy and ventricular enlargement. Hypofunction of NMDA receptors or their downstream effectors and/or the complement C4 risk gene represent two of the possible mechanisms responsible for the marked loss of cortical synapses. Third, NMDA receptor hypofunction impairs the cortical PV+-GABAergic neurons, which provide feedback inhibition to the pyramidal neurons and regulate cortical oscillations. Disinhibited cortical neuronal output drives increased subcortical dopamine release and psychosis. Thus, psychosis is a downstream consequence of the primary cortical pathology. Finally, the results of recent GWASs implicate more than 30 risk genes whose products are localized to synapses: clinical trials with agonists at the d-serine site on the NMDA receptor attenuate positive, negative, and cognitive symptoms. Translational studies with mice, in which NMDA receptor hypofunction has been genetically induced, confirm that the spotlight has moved from the dopamine system to glutamatergic circuits in schizophrenia. Nevertheless, this conceptualization accounts for the roles of only a portion of the identified risk genes. Hopefully, the not-too-distant future will bring us a much more complete understanding of the pathophysiology of schizophrenia and lead to more effective targeted treatments, making personalized medicine a reality for psychiatry.