Cardiovascular disease (CVD) is the leading cause of death in the United States, affecting over 650,000 people each year (
1). Rates of CVD and hypertension are even higher among individuals who have experienced trauma, and particularly those with posttraumatic stress disorder (PTSD) (
2,
3). PTSD is a highly impairing psychiatric condition with symptoms such as intrusions, avoidance of trauma-related stimuli, negative alterations in cognition and mood, and hyperarousal (
4). PTSD is often characterized by an exaggerated fear response controlled by the sympathetic nervous system. It is thought that chronic activation of this system strains the cardiovascular and immune systems, ultimately increasing susceptibility to CVD (
5). Despite the epidemiological evidence linking PTSD and CVD, there remain significant gaps with regard to how PTSD and CVD interact to confer risk in trauma-exposed individuals.
Given the preponderance of evidence supporting a PTSD-CVD link, the National Heart, Lung, and Blood Institute (NHLBI) convened a working group entitled “The Cardiovascular Consequences of Posttraumatic Stress Disorder,” which brought together experts from the NHLBI and the American Heart Association to determine research gaps and priorities (
6). As reviewed by O’Donnell et al. (
7), there are several gaps related to the genetics of PTSD and CVD: 1) “lack of large-scale datasets with deep genetic data and both PTSD and CVD measures”; 2) “lack of population cohort studies and biobanks to harness genetics and genomics to understand shared risks of PTSD and CVD”; and 3) “lack of polygenic risk scores from genome-wide association studies (GWASs) of PTSD and CVD to inform potential causal PTSD-CVD associations and identify potential therapeutic targets.” Addressing these gaps will improve PTSD and CVD risk identification, which can be used to improve PTSD treatment and reduce concurrent CVD risk. While there is still much to be done to address these gaps, the PTSD and CVD genetics fields have made significant advances with the advent of new technologies and more international collaboration.
Recent PTSD genetic studies have primarily taken a GWAS approach, catapulted by the creation of the PTSD division of the Psychiatric Genomics Consortium (PGC-PTSD) (
8). The first GWAS analyses in PTSD identified several risk genes and single-nucleotide polymorphisms (SNPs), including the rs8042149 SNP (
9), the Tolloid-like 1 gene (
10),
lincRNA AC068718.1 (
11), the
PRTFDC1 gene (
12), and the rs717947 SNP (
8,
13). In the first PGC-PTSD study, Duncan et al. (
14) found significant genetic overlap between PTSD and schizophrenia, and recent work identified sex- and ancestry-based differences in PTSD heritability (
15). The PTSD GWAS summary statistics from the PGC-PTSD have also been used to create polygenic risk scores (PRSs) that predict PTSD in large independent cohorts of veterans (
15,
16). Given the heterogeneity of PTSD and its polygenic nature, PRS studies using PGC-PTSD summary statistics will be crucial to improving our prediction of PTSD risk.
The field of CVD genetics has seen similar advances in terms of utilizing large samples across research groups. The Coronary Artery Disease Genome-Wide Replication and Meta-Analysis (CARDIoGRAM) consortium is comparable to the PGC-PTSD in that it was developed to allow for significantly larger studies with more power to detect genetic contributions to coronary artery disease (CAD; the largest subset of CVD) (
17). The consortium initially conducted a meta-analysis of GWASs and identified 13 risk loci for CAD, and several follow-up studies confirmed most of these and found evidence for additional loci implicating genes involved in vascular function (e.g.,
PECAM1,
PROCR) (
18–
20). GWAS has also been used to identify genetic risk for other aspects of CVD and its risk factors, such as blood pressure (
21), stroke (
22), atrial fibrillation (
23), carotid intima media thickness and carotid plaque (i.e., subclinical atherosclerosis) (
24), and heart failure (
25).
Three studies have reported on the shared genetics of PTSD and CVD. Using a candidate gene approach, Pollard et al. (
26) found that 37 PTSD risk genes relevant to immune function and inflammation were also independent risk genes for CVD. Sumner et al. (
27) used the PGC-PTSD GWAS summary statistics (
12) and found evidence for genetic overlap of PTSD and CAD, but these results did not survive correction for multiple testing. In a subsequent study, Sumner et al. (
28) used PGC-PTSD GWAS summary statistics and found significant main effects of PTSD symptoms for blood pressure in a trans-ethnic meta-analysis, although there was heterogeneity in the results. Given these promising findings, additional large-scale genetic analyses are needed to better account for the shared heritability of PTSD and CVD, and to improve genetic prediction. Further, it is critical to consider major depressive disorder (MDD) because it is highly comorbid with PTSD (
29), and like PTSD, it carries a significant risk of CVD (
30). Thus, genetic risk studies will be enhanced by considering PTSD, MDD, and CVD as inherently linked disorders.
The present study addresses gaps in the PTSD-CVD genetics literature as identified by the NHLBI working group (
6). We utilized a large-scale health care biobank data set (N=36,412) that contains CVD, PTSD, and MDD diagnostic data, as well as both PTSD and CVD GWAS summary statistics that included 1.3 million samples. Our goals were 1) to test genetic correlations between PTSD/MDD and CVD, and to estimate their magnitudes using current GWAS summary statistics data; 2) to leverage these relationships to improve genetic prediction performances; and 3) to identify the shared biological pathways or genes underlying the genetic correlations.
Methods
Participants and Procedure
This study included 36,412 participants from the Mass General Brigham Biobank (MGBB), which is a biorepository of Mass General Brigham Healthcare (e.g., Massachusetts General Hospital, McLean Hospital). Participants provide blood samples, complete health surveys, and consent to these being linked to clinical data from electronic health records (EHRs). This study included the subset of MGBB participants whose blood samples had been genotyped as of April 2020. PTSD diagnosis was determined based on the presence of at least one ICD-10 code in a patient’s EHR (i.e., based on clinical judgment). Hypertension and depression phenotypes were determined using “curated disease populations” determined using algorithms developed by the MGBB to increase phenotype accuracy (see the
online supplement) (
31,
32). Cardiovascular disease subphenotypes of congestive heart failure and ischemic stroke were also determined through curated disease populations, and myocardial infarction was determined based on the presence of at least one ICD-10 code. Pulse and average systolic and diastolic blood pressure were also collected from EHR data through the MGBB. Averages were determined from all of the measures of these vital signs available in individual health histories. Therefore, the number of collections included in the average differs for each patient. See
Table 1 for more information.
Genetic Data
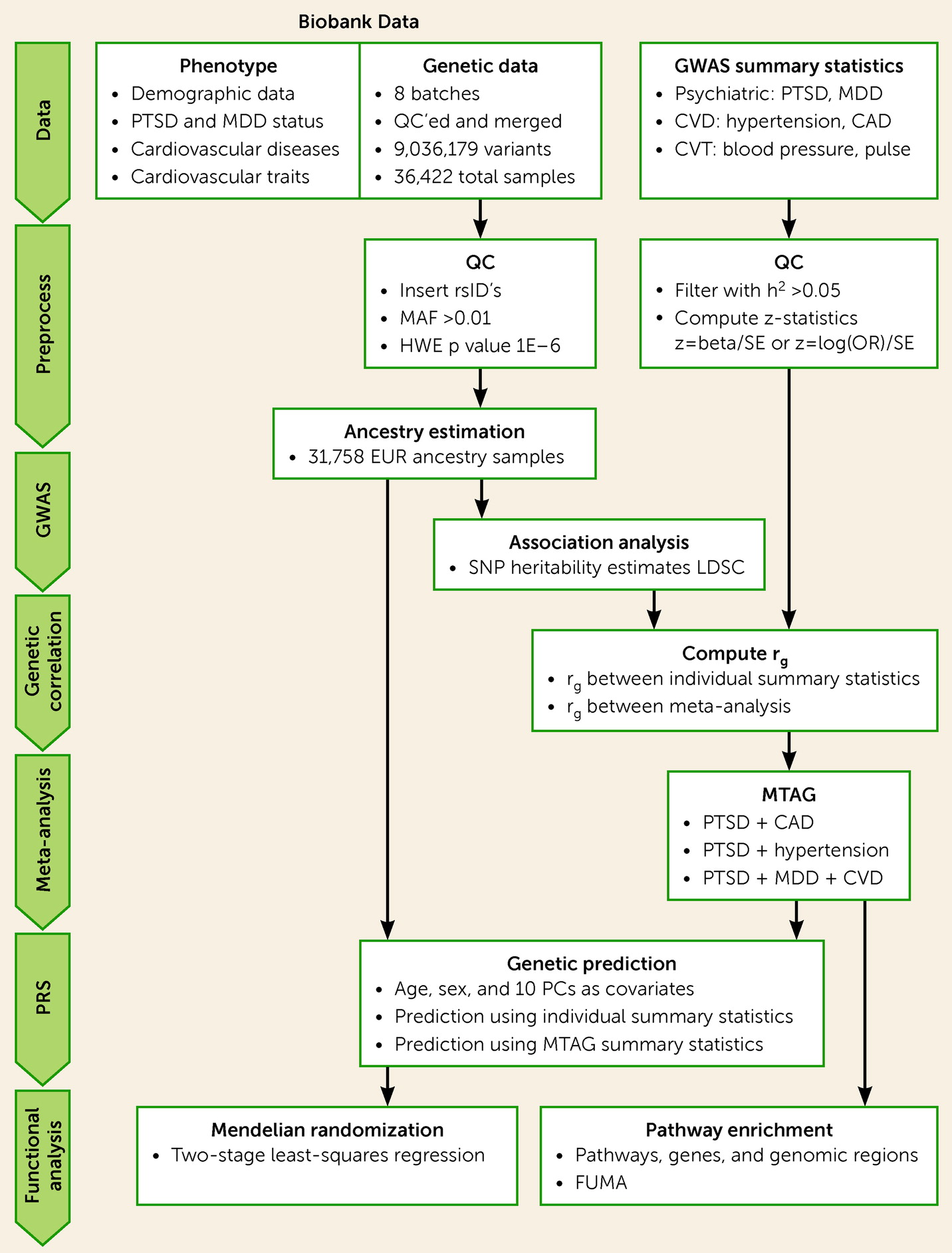
See
Figure 1 for an overview of the genetic analysis procedure. Briefly, de-identified genetic data were requested from the MGBB and were genotyped using Illumina SNP array. Quality control was performed on each batch individually and the batches were merged, resulting in a total of 36,422 individuals and 9,036,179 variants after imputation. Ancestry prediction was performed using a classification model trained on 1,000 Genomes samples (
39). Most participants included in the summary statistics are of European ancestry (>87%). To minimize the effect of population stratification, only those genetically predicted to have European ancestry (N=31,634) were included for further analysis. Self-identified and predicted ancestry sample sizes are listed in Table S1 in the
online supplement.
GWAS Summary Statistics Data
Publicly available GWAS summary statistics from several recent publications on PTSD, MDD, and CVD were collected. Summary statistics data sets were processed with the following quality control criteria: 1) SNP heritability h
2 >0.05; 2) compute z-statistics (z=beta/SE or z=log(OR)/SE); 3) filter nonvariable SNPs (0<freq<1); and 4) filter SNPs with low number of samples. Five summary statistics survived the quality control criteria and were used for further analysis: 1) ptsd_eur_freeze2: the PGC-PTSD freeze-2 (
15); 2) PGC_MDD2018_ex23andMe: the latest result from PGC-MDD excluding the 23andMe samples (
33); 3) UKBB_broad_Depression: UK Biobank (UKBB) analysis for depression phenotypes (
34); and large-scale genetic studies for 4) UKBB_Essential_Hypertension: hypertension (
35), and 5) CAD_Nikpay: CAD (
19). More detail on summary statistics is provided in Table S2 in the
online supplement.
For proprietary reasons, the PGC-MDD summary statistics that included the 23andMe samples are not publicly available. Therefore, we only used summary statistics conducted without the 23andMe cohort. With UKBB, summary statistics on three depression phenotypes were provided: probable MDD, ICD-defined MDD, and broad depression (defined by a self-reported help-seeking history for problems related to anxiety and depression) (
34). Only broad depression resulted in a SNP heritability estimate greater than 5%. Therefore, only broad depression summary statistics were included for further analysis.
GWAS and SNP Heritability Estimates on MGBB Data
We conducted a GWAS for four diseases: PTSD, MDD, hypertension, and CAD. A quantitative GWAS was also conducted for quantitative cardiovascular traits (average, minimum, and maximum pulse rate; systolic and diastolic blood pressure). Approximately 6 million variants were tested. Only variants on the 22 autosomal chromosomes were considered. To control for population stratification, the first 10 principal components were included as covariates. Age, sex, and batch were included as additional covariates. The analysis was performed using PLINK-2 (
36).
Combining Summary Statistics and Estimating Genetic Correlations
The multitrait analysis of GWAS (MTAG) approach was used to combine GWAS summary statistics (
37). MTAG is a generalization of inverse-variance weighted meta-analyses of summary statistics. However, unlike standard meta-analysis, MTAG computes trait-specific association analysis for each individual trait. Summary statistics for PTSD, MDD, hypertension, and CAD were incorporated.
Linkage disequilibrium (LD) score regression (LDSC) was used to estimate genetic correlations (
38). LDSC is a Python-based command line tool for estimating genetic correlations and SNP heritability from GWAS summary statistics. Precomputed LD scores for European ancestry from the 1,000 Genomes Project were used (
39). Genetic correlations were computed among individual summary statistics as well as among meta-analysis summary statistics for the four phenotypes (PTSD, MDD, hypertension, and CAD).
Constructing and Evaluating Polygenic Risk Score
PRS was computed as a weighted sum of the additively coded number of risk/effect alleles (0 for homozygous non-risk genotype, 1 for heterozygous genotype, and 2 for homozygous risk genotype). The weights were the beta or odds ratio from univariate test statistics obtained from the summary statistics of previous individual GWASs or with combined summary statistics using MTAG. Owing to methodological advantage (in particular, not requiring individual-level training genetic data), LD clumping followed by p-value thresholding is the popular way of selecting variants included in the score. The LD clumping step was done on a window of 250 kb and squared correlation of allele counts of 0.1. In the p-value thresholding step, PRS was computed for several thresholds. The optimal threshold is determined based on prediction performance as assessed by Nagelkerke’s R
2 (a pseudo R
2 for binary variables). Nagelkerke’s R
2 was computed as the difference of its value in the full model (which contains the PRS and covariates) and in the null model (which contains only the covariates). This is done for each of the PRSs independently. See Figure S1 in the
online supplement for results of the thresholding step. PRS analysis was conducted using PRSice-2 (
40), including age, sex, and the first 10 ancestry-related principal components as covariates.
To determine the prediction power of PRS, additional performance metrics were evaluated. Area under the receiver operating characteristic curve (AUC) was computed with the
pROC package in R (
41). Binary disease labels along with quantitative PRS values were used. To further evaluate the discriminatory ability of MTAG-PRS, stratification performance of the PRS was computed. This was done by binning samples into equal-sized risk strata based on PRS values. Next, disease incidence rate and odds ratio were computed for each risk group. For odds ratio, the first (the least-risk group) was used as the reference.
Mendelian Randomization
Mendelian randomization was used to test the causal link between PTSD and CVD (see the
online supplement for more information). Using two-stage least-squares regression, PTSD and MDD were our two exposure variables and hypertension and CAD were the two outcome variables, and the same models were tested in reverse with hypertension and CAD as the exposure variables and PTSD and MDD as the outcome variables (i.e., eight models tested). In the first stage, the exposure (binary PTSD or MDD label) is regressed against PTSD/MDD PRS and control variables (age and the first five principal components). Using this regression model, the predicted value of the exposure is computed. In the second stage, the outcome variable (binary hypertension or CAD label) is regressed against the predicted exposure variable and control variables. Next, epidemiological causality is inferred based on the significance of the coefficient of the predicted exposure variable.
Pathway Analysis
Pathway analysis was performed using FUMA (
42). FUMA is a web-based application that annotates a set of given variants from GWAS summary statistics by utilizing information from 18 data repositories and tools. It was implemented in two main analysis steps. In the first step (SNP2GENE), after taking a set of input variants, it added variants that were in linkage disequilibrium with the input significant variants, and SNPs were annotated (with biological functionality) and mapped to genes (with positional, eQTL, and chromatin interaction mapping). In the second step (GENE2FUNC), the set of prioritized genes from the first step were annotated with biological functions and mechanisms. This included enrichment in biological pathways and functional categories. Adjusted p values with Benjamini-Hochberg false discovery rate correction for multiple testing were computed.
Results
In the total sample, 2,149 participants were PTSD-positive case subjects and 34,269 were PTSD-negative control subjects. In terms of depression, 6,167 participants were predicted to have current MDD or a past history of MDD (of these, 1,133 had comorbid PTSD), while the remaining 30,243 were MDD negative. See
Table 1 for additional phenotypes and quantitative data by PTSD status.
Genetic Correlations Between PTSD and CVD
Genetic correlations were estimated in two ways. First, estimates were calculated using individual GWAS summary statistics (both publicly available as well as from new MGBB GWAS analysis). Then, large-scale meta-analysis summary statistics were computed by combining publicly available as well as the MGBB summary statistics for each of the four disease labels of primary interest. The four meta-analyses were PTSD (N=183,000): MGBB GWAS+PGC-PTSD freeze-2; MDD (N=362,584): MGBB GWAS+PGC-MDD+UKBB_ICD_MDD; hypertension (N=440,089): MGBB GWAS+UKBB_Essential_Hypertension; and CAD (N=217,000): MGBB GWAS+Nikpay_CAD. Using both of these approaches, statistically significant genetic correlations were estimated between PTSD and hypertension/CAD. With the meta-analysis summary statistics, PTSD demonstrated a significant positive genetic correlation with hypertension (r
G=0.35, SE=0.06, p=9.45E−10) and CAD (r
G=0.24, SE=0.06, p=5.09E−5). Similarly, MDD demonstrated significant genetic correlations with hypertension (r
G=0.31, SE=0.03, p=4.73E−33) as well as CAD (r
G=0.18, SE=0.03, p=8.99E−9). Genetic correlation estimates between PTSD/MDD and hypertension/CAD are listed in
Table 2.
Improving Genetic Prediction of PTSD
Next, we sought to leverage these robust correlations to improve the genetic prediction performances for PTSD diagnosis. First, we combined only publicly available summary statistics using MTAG. The MGBB samples were not included in the base/training summary statistics, which were used as the target/testing data set. As depicted in
Table 3 and
Figure 2, incorporating summary statistics for MDD and CVD resulted in improvement of both stratification and the discrimination ability of the PRS, such that the prediction of PTSD diagnosis was significantly enhanced. Using only PGC-PTSD freeze-2 summary statistics resulted in an AUC of 0.546 (95% CI=0.532, 0.560), while using summary statistics that combined PTSD, MDD, hypertension, and CAD resulted in an AUC of 0.592 (95% CI=0.578, 0.606). See Figure S2 in the
online supplement for hypertension and CAD prediction performance.
Mendelian Randomization to Determine Causal PTSD-CVD Link
Given that eight models were tested, we used a Bonferroni-corrected p value of 0.00625 (0.05/8). All of the models with PTSD and MDD predicting hypertension and CAD were significant using this threshold: PTSD predicting hypertension (β=0.20, SE=0.04, p=5.97E−6), PTSD predicting CAD (β=0.22, SE=0.07, p=9.22E−4), MDD predicting hypertension (β=0.43, SE=0.10, p=5.97E−6), and MDD predicting CAD (β=0.47, SE=0.14, p=9.22E−4). Of the models in the reverse direction, only the model with CAD predicting PTSD was significant, but barely over the threshold of p<0.00625 (β=0.31, SE=0.11, p=0.0041). See Table S4 in the
online supplement for a summary of all eight models. Overall, these findings provide greater support for a causal link in the direction from psychiatric disorders to hypertension and CAD, but not the reverse.
Pathway Enrichment Analysis With Shared Genetic Variants
A set of genetic variants at the significance threshold of p<1.0E−5 (for MTAG-combined GWAS results with MGBB along with base summary statistics that combined PGC-PTSD, UKBB-depression, hypertension, and CAD) were used to perform pathway annotation and functional analysis with FUMA. This p-value threshold was used to include more genes and variants for pathway analysis, which is a common practice for these analyses (
43). In the first step, SNP2GENE, the set of variants are mapped to genes based on genetic coordinates. The 87 genomic risk loci included in the pathway analysis are shown in Figure S3 in the
online supplement. The 8,734 SNPs falling in these genomic risk loci are mapped to 198 genes. In the second step, GENE2FUNC, the prioritized genes are used as input to the second step to perform functional annotation. Top significantly enriched gene ontology biological processes in the set of genes included those involved in the regulation of postsynaptic structure (p=0.00001, p
adj=0.005), synapse organization (p=0.0001, p
adj=0.02), and interleukin-7 (IL-7)–mediated signaling pathways (p=0.0002, p
adj=0.03). In addition to the pathways, tissue specificity was assessed using differentially expressed genes (comparing a given tissue label with all other labels). However, no statistically significant result was found (see Figure S4 in the
online supplement).
Discussion
This is the first study to examine the shared genetic susceptibility of PTSD and CVD utilizing large-scale genome-wide cross-trait analysis from both PTSD and CVD GWAS summary statistics, which were derived from a total of 1.3 million samples, and 31,758 genotyped samples that were used as the target/test data set. We identified significant genetic correlations between PTSD and CVD, as well as MDD, and Mendelian randomization analyses provided support for a causal link from PTSD to hypertension and CAD, but not the reverse. We were also able to significantly improve the genetic prediction of PTSD by incorporating summary statistics from CVD and MDD. Further, pathway enrichment analyses indicated that genetic variants involved in shared PTSD-CVD risk included those involved in postsynaptic structure, synapse organization, and immune function. These findings suggest that PTSD and CVD risk identification may be improved by their mutual consideration, which can be used to improve PTSD treatment selection.
A preponderance of epidemiological data has suggested that individuals with PTSD are more likely to experience CVD (
2,
3). As identified by the NHLBI working group (
6) on PTSD-CVD risk, large-scale studies are needed to better understand the shared genetic risk of these diseases. Here, we provide the first evidence of shared genetic risk for PTSD and CVD using summary statistics from both literatures. We also found significant genetic associations of PTSD and CVD with MDD, which is consistent with previous research supporting comorbidity of MDD with both PTSD and CVD (
29). While the overlap of mental disorders is increasingly accepted in psychiatry (
44), our results suggest that two highly impairing psychiatric conditions also overlap with a set of cardiovascular diagnoses thought to have more discrete origins. This provides further evidence that psychiatric and physical health conditions are not only common in phenotypic presentation, but they are also genetically linked.
Our Mendelian randomization analyses found evidence that genetic risk for PTSD and MDD exerted a causal effect on hypertension and CAD risk. One of the primary mechanisms implicated in the link between PTSD/MDD and CVD is elevated sympathetic arousal that leads to hypertension, both directly (i.e., chronically elevated blood pressure due to stress) and via the renin-angiotensin system (i.e., elevated blood pressure due to renin and angiotensin-II release that causes vasoconstriction). Our findings provide support for this mechanism, but we cannot exclude the possibility that confounding factors or mediating effects (e.g., diet, smoking) are responsible for these associations, which should be examined in future research. Our pathway enrichment analyses suggested that genetic variants involved in postsynaptic structure, synapse organization, and immune function (IL-7) may be relevant to the shared genetic risk of PTSD and CVD. While IL-7 has not previously been studied in the context of PTSD, other immune markers and genes (e.g., IL-6, IL-8) have been implicated in PTSD, and it has been suggested that impaired immune function may be a mechanism underlying the PTSD-CVD link (
5,
26). Given that IL-7 has been implicated in hypertension and atherosclerosis (
45,
46), this further implicates the role of immune function in the shared genetic risk of PTSD and CVD, and it suggests that additional study of these genetic variants is warranted.
The primary clinical implication of this work is improved risk identification, which can be used to enhance PTSD treatment and decrease CVD risk. For example, identifying individuals with shared PTSD and CVD risk (and knowing that PTSD is a specific risk factor for CVD) will allow clinicians to select PTSD interventions that are known to improve aspects of cardiovascular function. There is preliminary evidence that forms of cognitive-behavioral therapy (the first-line treatment for PTSD) improve cardiovascular function (
47), but this research is still in its early stages. On the other hand, psychiatric and CVD medications have well-documented effects on cardiovascular function (
48,
49), but few studies have tested the effects of these medications on subsequent CVD risk in PTSD populations. Medications targeting the renin-angiotensin system (e.g., ACE inhibitors, beta-blockers) have demonstrated efficacy in rodent models, but research in humans with PTSD has been mixed (
50,
51). A next step in this line of work is to determine whether existing cognitive-behavioral and pharmacological treatments actually reduce CVD risk in PTSD, and to determine whether they are more efficacious for individuals with high genetic risk for both PTSD and CVD. An additional and more immediate next step is to include PTSD as an indicator of CVD risk in routine care (e.g., during hypertension screening).
While our use of a large biobank sample is a strength of this study, it also has inherent limitations. For example, PTSD diagnosis was based on single ICD-10 codes, which were not necessarily the result of standardized assessments (e.g., the Clinician-Administered PTSD Scale [
52]). This approach, while necessary for large-scale data analyses, is not as reliable as using clinician-administered assessments of PTSD, as we cannot determine whether some PTSD diagnoses were given incorrectly. Another limitation is that the MGBB sample comprises primarily White, non-Hispanic individuals. As a result, over 87% of participants were of European ancestry, and analyses were conducted only on these individuals. While this approach minimized the effect of population stratification, it means that our findings may not replicate to other racial/ethnic/ancestry groups. Finally, it is important to note that the relationship between PTSD and CVD is likely mediated, at least in part, by behavioral risk factors, such as diet, exercise, smoking, and alcohol/substance use. These risk factors are independently associated with CVD risk and cardiometabolic diseases that are linked with CVD (e.g., type 2 diabetes). Thus, a limitation of this study is the lack of data on these behavioral variables that likely influence the pathways through which PTSD may lead to CVD. Research using multivariate Mendelian randomization approaches is needed to further assess the role of possible confounding or mediating factors in the PTSD-CVD link. Similarly, it will be critical for future studies to test whether the PTSD and MDD correlations with CAD depend on hypertension, and whether PTSD and MDD each has unique risk associated with hypertension and CAD independent of the other.
The link between PTSD and CVD has been well established from an epidemiological standpoint. As identified by the 2018 NHLBI working group (
6), large-scale genetic studies are needed to improve characterization of PTSD and CVD risk, which will subsequently improve risk management and treatment. We directly addressed some of these needs by utilizing a large biobank sample (N=36,412) and by leveraging large-scale GWAS summary statistics from both the PTSD and CVD literatures with 1.3 million samples. Our results indicate that 1) there is substantial genetic overlap between PTSD and CVD, 2) PTSD and MDD may be risk factors leading to the development of hypertension and CAD, and 3) the genetic prediction of PTSD risk is improved by the consideration of polygenic risk for CVD and MDD. Future studies of genetic risk and diagnostic prediction would benefit from incorporating this polygenic risk approach.