Several lines of evidence from recent studies with blood or urine or with magnetic resonance spectroscopy support the idea that mitochondrial dysfunction is implicated in the pathophysiology of ASD (
3,
4). Moreover, while the mitochondrial electron transport chain complex I (MC-I) genes are encoded in both the nuclear and mitochondrial genomes, postmortem studies of autistic brains have found lower nuclear gene expression of MC-I (
5,
6). MC-I is the first rate-limiting step of the electron transport chain, which operates to produce energy in the form of ATP; this is responsible for neurotransmitter synthesis and synaptic plasticity (
7). Because the brain has a high ATP demand, the finding of decreased MC-I indicates a role of mitochondrial dysfunction in the brain pathophysiology of ASD. More specifically, postmortem studies have found altered MC-I in the anterior cingulate cortex (ACC), superior temporal gyrus, occipital cortex, dorsolateral prefrontal cortex, thalamus, and primary motor cortex of individuals with ASD (
5,
6,
8). However, no previous studies have examined mitochondrial dysfunction in the living brains of individuals with ASD, or its relationship with the core symptoms of ASD.
In vivo estimates of MC-I availability in the living human brain are now possible using positron emission tomography (PET) measurements with 2-
tert-butyl-4-chloro-5-{6-[2-(2-[
18F]fluoroethoxy)-ethoxy]-pyridin-3-ylmethoxy}-2H-pyridazin-3-one ([
18F]BCPP-EF), a radioligand that binds to MC-I and permits investigation of the topographical distribution of mitochondrial dysfunction (
9). Our previous studies confirmed that the uptake of [
18F]BCPP-EF reflected the specific binding to cellular MC-I (for details, see the
online supplement). By utilizing [
18F]BCPP-EF PET, the present study aimed 1) to investigate whether and where mitochondrial dysfunction occurs in the living brains of individuals with ASD, and 2) to identify the clinical correlates of detected mitochondrial dysfunction.
Discussion
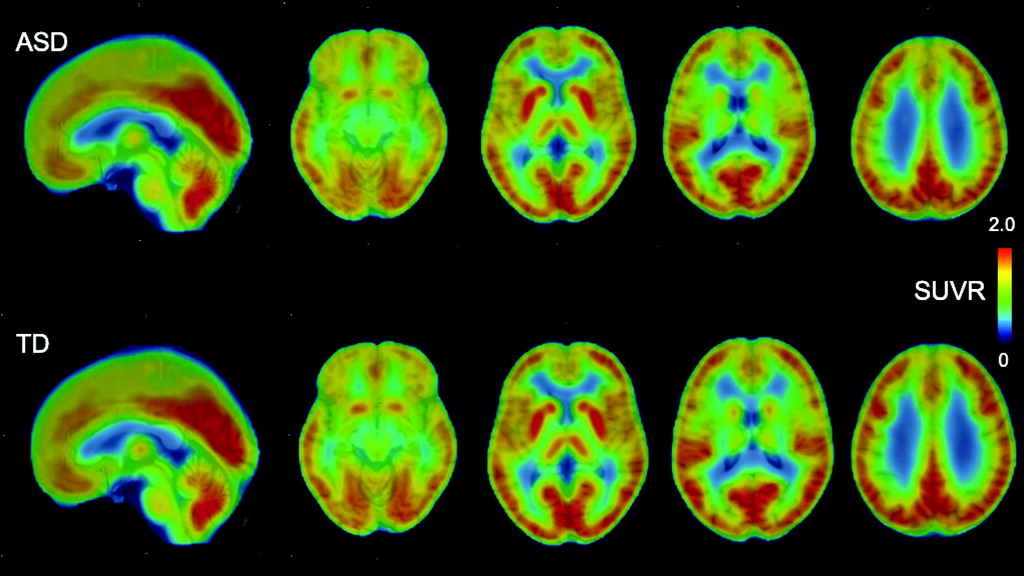
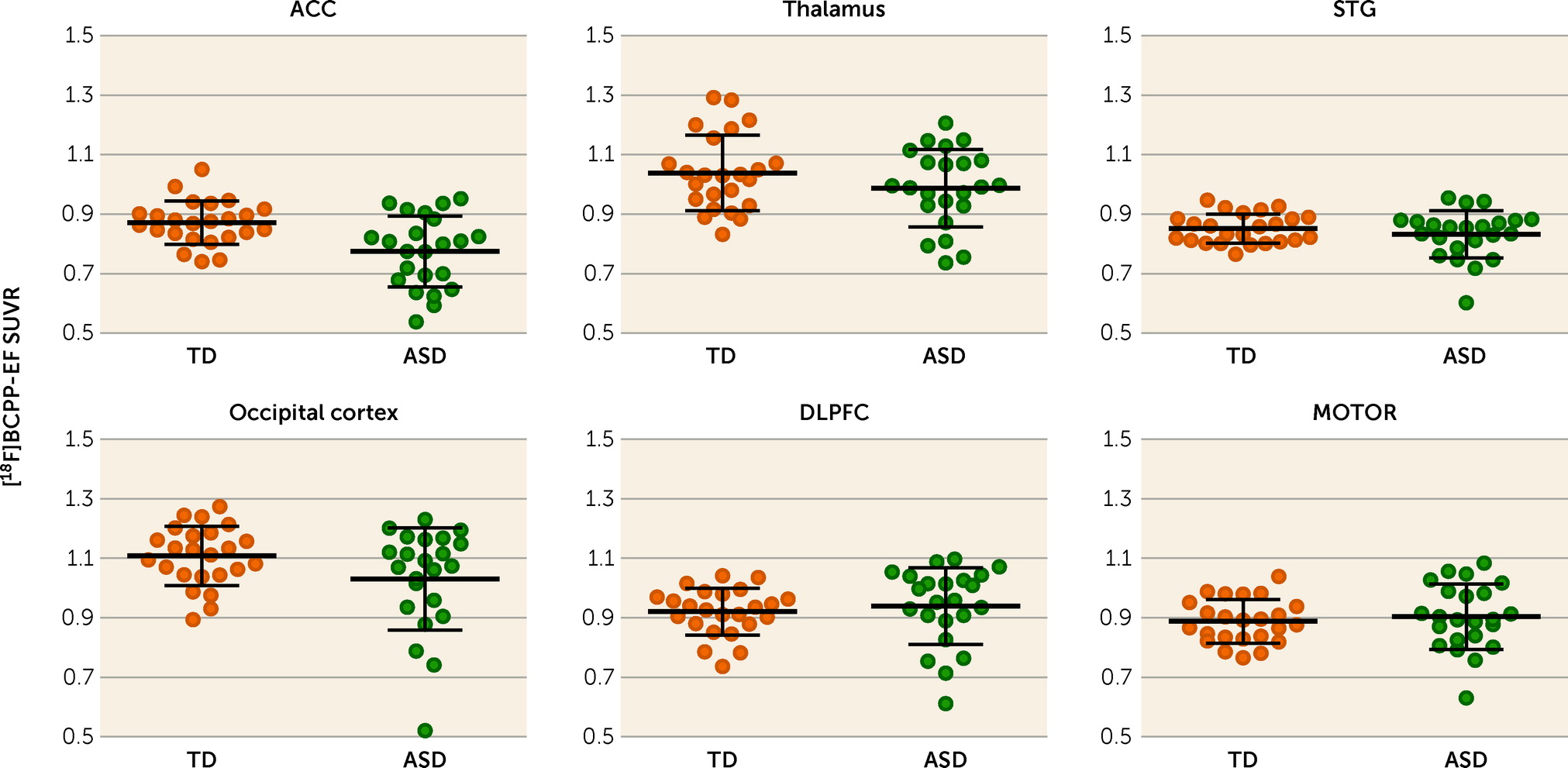
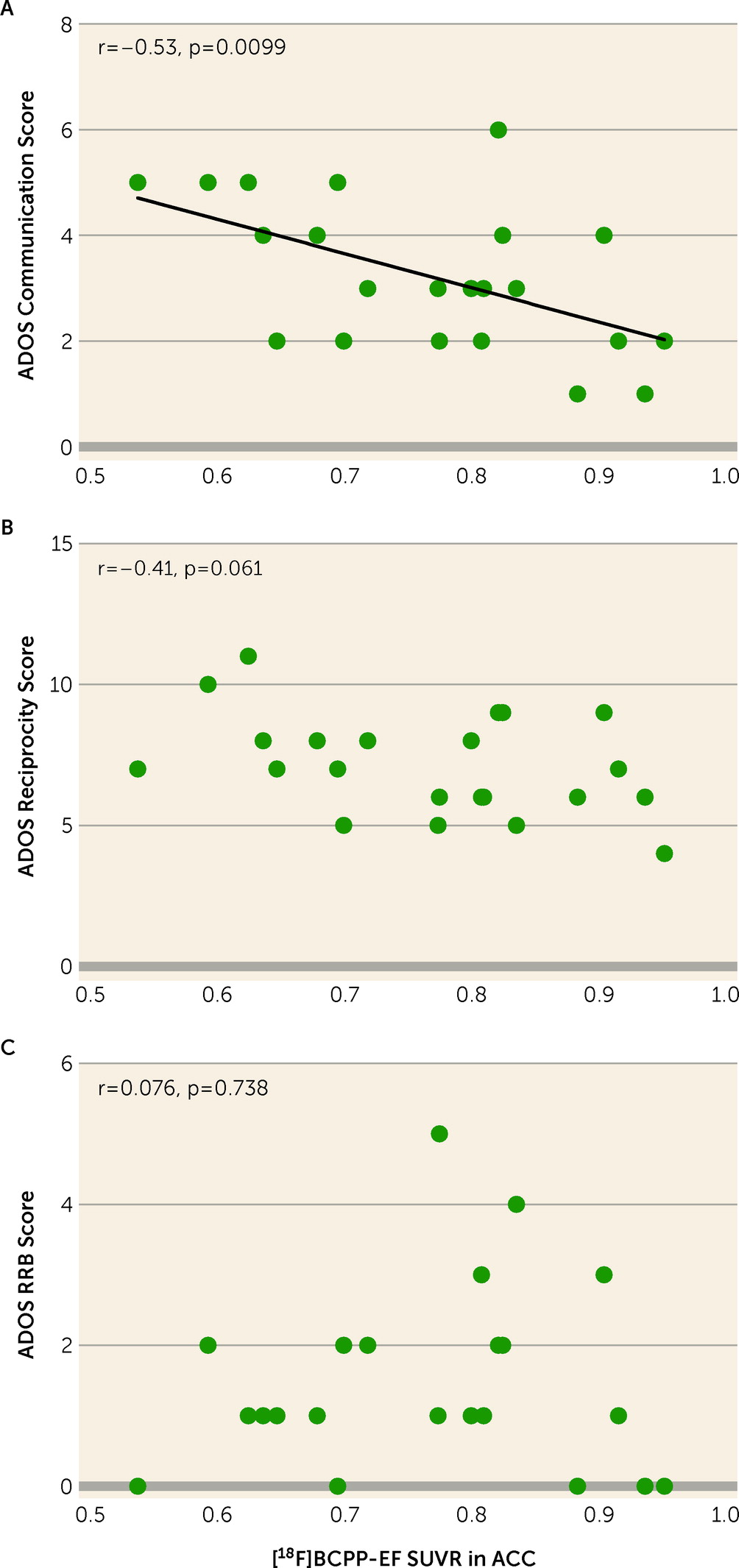
To the best of our knowledge, the present study demonstrates for the first time lower MC-I availability in the ACC of psychotropic medication–free and mitochondrial disease–free adult males with high-functioning ASD compared with demographically matched typically developed individuals. The decreased availability of MC-I in the ACC of participants with ASD was significantly correlated with the severe ADOS-2 communication score. As revealed in our previous studies, the decreased [18F]BCPP-EF binding indicates brain regional mitochondrial dysfunction, including the degradation and quantitative reduction of MC-I proteins as well as reduced functional activities of MC-I (for details, see the online supplement).
The low availability of MC-I in individuals with ASD compared with typically developed individuals is consistent with previous reports suggesting a critical role of mitochondrial dysfunction in the pathogenesis of ASD. A meta-analysis of peripheral biomarkers (
4) showed the high prevalence of mitochondrial disease and abnormal values in lactate, pyruvate, alanine, creatine kinase, ammonia, and aspartate aminotransferase in the general population of individuals with ASD. Reduced mitochondrial complex activities and protein levels in postmortem brain (especially in the frontal cortices, including the ACC) and in peripheral samples have repeatedly been reported in conjunction with alterations in MC-I in individuals with ASD (
3,
4,
18). A downregulation in MC-I genes in the postmortem autistic brain and associations between mitochondrial DNA variants and ASD have also been reported (
3,
5,
19). The elevated production of reactive oxygen species, which are produced during the metabolism of oxygen at the electron transport chain including MC-I, and increased markers of oxidative stress were reported in individuals with ASD in another meta-analysis (
20). Moreover, magnetic resonance spectroscopy studies investigating brain lactate have suggested abnormalities in mitochondrial energy metabolism (
21,
22). In addition, previous postmortem studies as well as in vivo PET studies have reported the abnormal expression of translocator protein, which is an activated glial marker that is expressed on mitochondrial external membranes, and have suggested mitochondrial dysfunction in the autistic brain (
16,
23,
24). Translocator protein has been implicated in several physiological processes, including immune modulation and mitochondrial homeostasis, while BCPP specifically binds to MC-I and reflects mitochondrial function regardless of immune modulation. Our results support these previous studies implicating mitochondrial dysfunction, and specifically lower MC-I, in the pathophysiology of ASD, and further extend the evidence for mitochondrial dysfunction in ASD by revealing lower levels of MC-I in the living brain for the first time.
In line with the present finding of decreased MC-I availability in the ACC of individuals with ASD, previous studies have supported an important role of the ACC in the pathophysiology of ASD. Functional MRI studies have repeatedly revealed dysfunction of the ACC in individuals with ASD (
25). Additionally, a lower density of myelinated axons and a decreased expression of axon guidance receptors have been identified in postmortem brains of individuals with ASD (
26). Because mitochondria regulate axon guidance receptors, decreased MC-I activity may lead to malfunctions in axonal outgrowth (
27), and the ensuing disorganization in axon guidance may disrupt functional and structural connectivity in individuals with ASD (
28). The present study further offers direct evidence for lower MC-I activity in the ACC in vivo in individuals with ASD.
Although previous postmortem studies have reported decreased nuclear gene expression of MC-I in all of the ROIs that we investigated, our study detected a significant group difference in MC-I availability specifically in the ACC. This discrepancy might be explained by the potential confounding factors of postmortem studies. In postmortem studies, the major causes of death in individuals with ASD were drowning, asphyxiation, and seizure, which led to a hypoxic event (
5,
6,
13). Hypoxia is reported to deactivate the MC-I (
29), while seizure activity impairs mitochondrial energy production by affecting the activity of mitochondrial enzymes (
30). Furthermore, previous reports have suggested that antidepressants, antipsychotics, and selective serotonin reuptake inhibitors impair MC-I activity and ATP production (
31,
32). All of these factors might therefore affect the results of postmortem studies (
5,
6,
13). In contrast, because no participant in the present study was taking psychotropic medication or had a seizure history, it is unlikely that these factors confounded the study results.
The significant correlation between reduced MC-I activity in ACC and high ADOS-2 communication scores suggests that mitochondrial dysfunction in ACC plays an important role in ASD social communication deficits. Previous neuroimaging studies have also supported a significant contribution of the ACC to ASD social communication deficits. Based on hypoactivation of the ACC in response to social rewards in ASD (
25), the social motivation theory suggests that individuals with ASD experience less reward for social interactions than do typically developed individuals (
33). Reduced glucose metabolism in the ACC, as assessed by
18F-FDG PET, which is a well-established technique for the quantitative measurement of the regional metabolic rate of glucose in the brain and reflects glucose metabolism as well as neuroinflammation, has also been reported to correlate with more severe ADI-R social domain deficits (
34). PET imaging with
18F-BCPP-EF, which has specificity for MC-I activity and which is not affected by inflammation with microglial activation, could be expected to provide more accurate information about neurodevelopmental pathophysiology than
18F-FDG. Moreover, levels of
N-acetylaspartate, which is synthesized in mitochondria and reflects MC-I activity, are correlated with ADOS-2 social affect scores in individuals with ASD (
35). In the ACC of postmortem ASD brains, the dysregulation of excitatory neuron-related genes was reported to correlate with total ADI-R score (
36). Our results in the present study further suggest that MC-I dysfunction is a candidate mechanism underlying the role of the ACC in autistic social communication deficits.
The finding of MC-I dysfunction in the present study signals its possible importance as a novel therapeutic target for ASD. A randomized trial showed that folinic acid, which is one of the vitamins that are needed for MC-I activity (
37), improves verbal communication in children with ASD (
38). Furthermore, several studies have suggested that the ACC, where we found mitochondrial dysfunction, is associated with the neural processes underlying therapeutic mechanisms in ASD, such as improved social behaviors with genetic activation of SHANK3 in a mouse model having dysfunctional pyramidal neurons in the ACC (
39), improved activity and functional connectivity in the ACC with oxytocin administration (
40,
41), and modified neural plasticity associated with the
N-methyl-
d-aspartate receptor (NMDAR) induced by NMDAR agonists or oxytocin (
42,
43). The present study provides further evidence to support the contribution of MC-I dysfunction in the ACC to the core symptoms of ASD, thus indicating its potential as a novel therapeutic target.
Some methodological considerations and limitations of the present study must be borne in mind. First, the participants were restricted to high-functioning young adult Japanese males, free of psychotropic medication. Although the uniformity of demographic characteristics enhanced our ability to detect the difference in MC-I availability in the ACC of ASD participants and typically developed participants with a large effect size (d=0.98), care should be taken when generalizing the study findings. The effect of MC-I availability might be found to be smaller in other ASD populations, such as females, medicated individuals, and children. Second, although the target sample size of our study was determined on the basis of previous PET studies in ASD (
15–
17), a future study with a larger sample size might detect significant associations between MC-I availability and other core symptoms of ASD that had marginal significance in the present sample (for example, the association with ADOS-2 reciprocity score approached significance at p=0.061; see Table S2 in the
online supplement). Third, our study was a cross-sectional comparison, and we are therefore unable to infer causality for the role of MC-I availability in the pathophysiology of ASD. Fourth, this study did not evaluate regional cerebral blood flow. Although previous studies have shown that [
18F]BCPP-EF availability was not influenced by regional cerebral blood flow in the conscious monkey brain (
44,
45), the possibility that regional blood flow affects [
18F]BCPP-EF availability in the human brain cannot be ruled out. Fifth, given the difficulty associated with the placement of an invasive arterial line that would allow assessment of volumes of distribution in participants with ASD, in the present study, we utilized the surrogate SUVR measure instead of volumes of distribution. Although our previous studies have shown correlations between volumes of distribution and SUVR, these were not in a disease-specific manner (
46,
47), which casts doubt on whether we could make any inferences regarding high-functioning ASD. Sixth, to minimize potential confounding effects, we recruited participants who were not taking any psychotropic medication and did not have any psychiatric comorbidities, and we screened their peripheral blood to rule out mitochondrial diseases. Even with these steps, we cannot rule out medical comorbidities that might increase the chances of finding mitochondrial dysfunction.
In conclusion, this study provides the first evidence from in vivo human brain of lower MC-I binding in the ACC of individuals with ASD compared with typically developed individuals, and of its relationship to severe social communication deficits. These findings indicate an important role of MC-I in the pathophysiology of ASD and suggest the potential of MC-I as a novel molecular target for pharmacological treatments of ASD core symptoms.