Major depressive disorder is a serious, disabling, life-shortening illness with a high lifetime risk: 7%–12% for men and 20%–25% for women (
1). It is often recurrent, episodes frequently last more than 2 years (i.e., are chronic) (
2), and interepisode recovery is often incomplete (
3). Chronic episodes and recurrent courses are associated with worse prognoses and are more likely to need longer-term treatment (
4–
7).
Could remission rates be increased with a combination of two antidepressant medications used together as initial treatment? Other branches of medicine often employ combination treatments at the outset of chronic illness, especially for the more severely ill (
9,
10). In depression treatment, when a single antidepressant medication is not effective (
7,
11), a second is often added to the first, with some evidence for efficacy (
12). It also appears that some antidepressant medications work for some patients but not for others. A combination of two medications might therefore increase the spectrum of patients who could benefit from the combination (
13). Furthermore, an unexpected synergy between medications might produce a rapid onset of benefit, so that fewer patients would drop out of treatment, which, in turn, might enhance remission rates. From a pharmacological perspective, a combination might affect a wider range of neurotransmitter or neuromodulator systems, which would enhance efficacy for some patients (
14–
16). Finally, clinical experience and a few small randomized, short-term trials (
13,
17,
18) suggest that some combinations can be more effective than monotherapy. On the other hand, treatment guidelines do not recommend such an approach as a first treatment step, and the risk of serious adverse events or intrusive side effects has not been fully evaluated. Thus, combining antidepressants as a first-step treatment for depression needs proper evaluation.
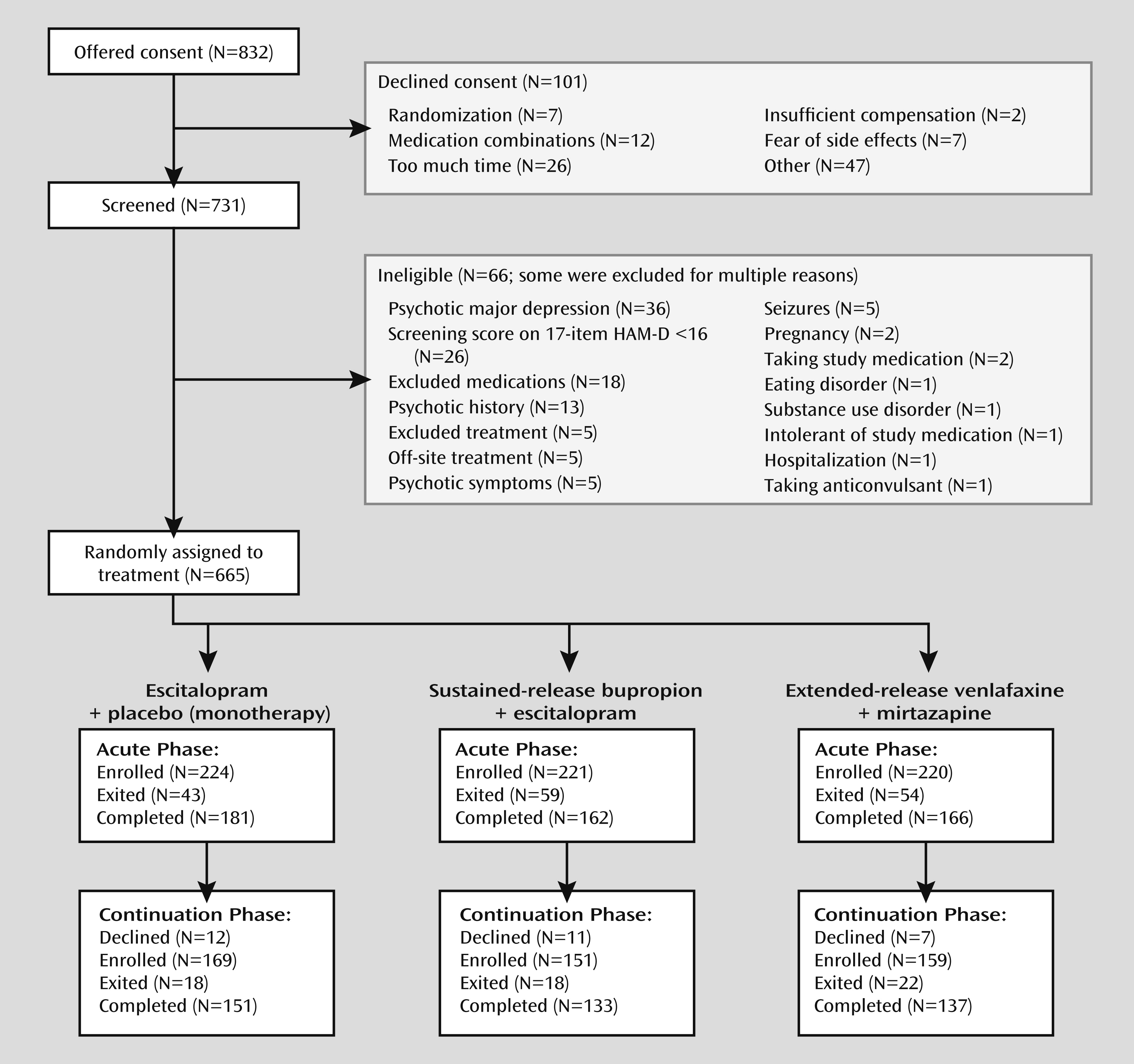
The Combining Medications to Enhance Depression Outcomes (CO-MED) trial was designed as a proof-of-concept study to determine whether either of two different antidepressant medication combinations would produce a higher remission rate at 12 weeks and, secondarily, after 7 months than monotherapy with a selective serotonin reuptake inhibitor (SSRI) as a first-step treatment in outpatients with chronic or recurrent major depression. We also compared the treatment effects on patient retention, side effect burden, and quality of life.
Method
Study overview
CO-MED was a 7-month single-blind, randomized, placebo-controlled trial that compared the efficacy of each of two medication combinations with escitalopram plus placebo in a 1:1:1 ratio as first-step treatment, including acute-phase (12 weeks) and long-term continuation (total 7 months) treatment. We planned a study group of 660 outpatients with nonpsychotic major depression from six primary and nine psychiatric care sites to allow detection of roughly a 15% difference in remission rates between each combination and escitalopram-placebo (with an expected remission rate of 35%). This difference was viewed as sufficiently large to affect practice since it approximates the benefit of a single antidepressant medication over placebo in successful antidepressant registration trials (
8).
Site selection
Clinical sites were selected on the basis of our prior experience and their performance in the Sequenced Treatment Alternatives to Relieve Depression trial to ensure 1) adequate patient flow, 2) committed administrative support, 3) adequate minority representation, and 4) adequate representation of both primary and psychiatric care sites.
Recruitment
Potential participants were screened at each clinical site with each site’s standard procedure (variable across sites). Most sites used two to nine questions from the Patient Health Questionnaire (
19,
20). Patients identified by screening saw their study clinicians and clinical research coordinator to determine study eligibility following written informed consent.
Participants
Broad inclusion and minimal exclusion criteria ensured a reasonably representative participant group. The outpatient enrollees were 18–75 years old and met the DSM-IV-TR (
21) criteria for either recurrent or chronic (current episode lasting at least 2 years) major depression according to a clinical interview and confirmed with a DSM-IV-based symptom checklist completed by the clinical research coordinator. Eligible participants had to have an index episode lasting at least 2 months and had to score at least 16 on the 17-Item Hamilton Depression Rating Scale (HAM-D) (
22). Those with any psychotic illness or bipolar disorder and those in need of hospitalization were ineligible. (For a complete list of exclusion criteria, see
http://clinicaltrials.gov/ct2/show/NCT00590863.)
The study protocol and all consent and study procedures were approved by the institutional review boards at the national coordinating center (University of Texas Southwestern Medical Center at Dallas), the University of Pittsburgh data coordinating center, and each participating regional center and relevant clinical site.
Baseline data
Sociodemographic and illness features were recorded at baseline. The anxiety subscale of the HAM-D was used to establish the presence of anxious features at baseline (
23). This anxiety/somatization factor, derived from a factor analysis of the HAM-D conducted by Cleary and Guy (
24), includes six items from the original 17-item version: item 10 (anxiety, psychic), item 11 (anxiety, somatic), item 12 (somatic symptoms, gastrointestinal), item 13 (somatic symptoms, general), item 15 (hypochondriasis), and item 17 (insight). A HAM-D anxiety/somatization factor score of 7 or higher indicated anxiety. The HAM-D administered at baseline by research outcome assessors (not located at any clinical site) was used to define anxious features. The self-report Psychiatric Diagnostic Screening Questionnaire (
25) was used to establish the presence of current axis I disorders. The Self-Administered Comorbidity Questionnaire (
26) established the presence, severity, and functional impact of a range of common concurrent general medical conditions.
Antidepressant treatment
A 12-week study period was chosen for the primary analysis to provide sufficient time for maximal dosing (if needed) and to allow most cases of depression that could remit to do so (
27). Treatment visits were planned at baseline and weeks 1, 2, 4, 6, 8, 10, 12, 16, 20, 24, and 28. No washout was required, but clinicians could choose a washout period if they thought it to be clinically advisable. Dose adjustments were based on measurement-based care following an operations manual to provide personally tailored but vigorous dosing (
28). Dose adjustments were based on the score on the 16-item Quick Inventory of Depressive Symptomatology—Clinician-Rated (QIDS-C) (
29), which was extracted from the 30-item Inventory of Depressive Symptomatology—Clinician-Rated (IDS-C) (
30), and the score on the Frequency, Intensity, and Burden of Side Effects Rating Scale (
31) obtained at each visit.
Treatment was randomly assigned, stratified by clinical site according to a web-based randomization system (
32). Random block sizes of three and six were used to minimize the probability of identifying the next treatment assignment. Dosing schedules were based on prior reports (
33–
35). Doses were increased only in the context of acceptable side effects. Participants could exit the study if unacceptable or intolerable side effects occurred and could not be resolved with dose reduction or medication treatment of the side effects.
Escitalopram plus placebo (monotherapy)
Escitalopram treatment was begun at 10 mg/day (one pill) and could be increased to 20 mg/day (two pills) at 4 weeks if the score on the QIDS-C was higher than 5 (if side effects allowed). Pill placebo was started at week 2, with the option to increase it to two pills at week 4 if the QIDS-C score was higher than 5 (if side effects allowed).
Bupropion plus escitalopram
The dose of sustained-release bupropion was 150 mg/day initially and was increased to 300 mg/day at the week 1 visit. Escitalopram was begun at 10 mg/day at the week 2 visit. At week 4, the bupropion dose was raised to 400 mg/day (200 mg b.i.d.) and/or the escitalopram dose was raised to 20 mg/day if the score on the QIDS-C was higher than 5 (if side effects allowed). At week 6 and beyond, doses could be increased up to a maximum of 400 mg/day (200 mg b.i.d.) for bupropion and 20 mg/day for escitalopram if the QIDS-C score was above 5 (if side effects allowed).
Venlafaxine plus mirtazapine
Treatment with extended-release venlafaxine was begun at 37.5 mg/day for 3 days and then raised to 75 mg/day. At week 1, the dose was raised to 150 mg/day. At week 2, if the score on the QIDS-C was above 5, mirtazapine was added at a dose of 15 mg/day. At week 4, if the QIDS-C score was above 5, the venlafaxine dose was raised to 225 mg/day and/or the mirtazapine dose was increased to 30 mg/day. At week 6, if the QIDS-C score was higher than 5, the mirtazapine dose could be raised to 45 mg/day, the maximum dose. At week 8, if the score was above 5, the venlafaxine dose could be raised to 300 mg/day, the maximum allowed.
Medication blinding
The first medication given in each treatment group was open label (both participant and study personnel were unblinded), while each second medication was given in a single-blind fashion (participant only) to ensure that the participants all took two types of pills. Specifically, in the escitalopram-placebo cell, placebo administration was blinded. For the bupropion-escitalopram combination, escitalopram was blinded. For venlafaxine-mirtazapine, mirtazapine was blinded. The participants remained blinded to the second medication throughout the 7-month study. The research coordinators and physicians were not blinded, to maximize safety and facilitate informed flexible dosing decisions.
Concurrent treatments
Only protocol antidepressant medications were allowed. Treatments with possible antidepressant effects were proscribed, as were anxiolytics, sedative-hypnotics, and depression-targeted, empirically validated psychotherapies for depression. Other therapies (e.g., supportive, couples, occupational) were allowed, as were medications for any general medical condition. Given the inhibition of the 2D6 isoenzyme by sustained-release bupropion, we alerted clinicians about nonprotocol medications (e.g., type 1C antiarrhythmics, beta-blockers) for which serum or dose adjustments might be needed. Medications to treat antidepressant medication side effects were allowed; administration was based on clinician judgment.
Research outcomes
Outcome measures were assessed at baseline and all treatment visits. The primary outcome, symptom remission, was based on the score on the 16-item Quick Inventory of Depressive Symptomatology—Self-Report (QIDS-SR) at 12 weeks (
29). The designation of remission was based on the last two consecutive measurements during the 12-week acute-phase trial to ensure that a single “good week” was not falsely signaling remission. At least one of these ratings had to be less than 6, while the other had to be less than 8. If participants exited before 12 weeks, their last two consecutive scores were used to determine remission. Those without two post-baseline measures were considered not to have remission.
Physicians were advised that participants could exit the study if they had received a maximally tolerated dose(s) for 4 or more weeks by week 8 without obtaining at least a 30% reduction from the baseline score on the QIDS-C. They could enter continuation treatment (weeks 12–28) if they had received an acceptable benefit (defined as a QIDS-C score of 9 or less by week 12) or if they reached a score of 10–13 and the clinician and participant judged the benefit to be substantial enough to recommend treatment continuation. Thus, virtually all participants entering the continuation phase had at least a 40% reduction in the QIDS-C score. If a participant exited the study at any time, a study exit form was completed. Clinical research coordinators attempted to contact all participants who did not come for a final exit visit.
Secondary outcomes included attrition, anxiety as reflected in the score on the anxiety subscale of the IDS-C (
30), functioning as measured by the Work and Social Adjustment Scale (
36), quality of life as measured by the Quality of Life Inventory (
37), side effect burden as measured by the Frequency, Intensity, and Burden of Side Effects Rating Scale (
31), and specific side effects as measured by the Systematic Assessment for Treatment Emergent Events—Systematic Inquiry (
38). Manic symptoms were assessed by using the Altman Self-Rating Mania Scale (
39), and cognitive and executive dysfunction was assessed by means of the Cognitive and Physical Functioning Questionnaire (
40).
Statistical analyses
Descriptive statistics, including measures of central tendency and dispersion, were computed for continuous data. Frequency distributions were estimated for categorical data.
Outcome analyses were conducted with the full group, on the basis of the intention-to-treat principle. Each combination therapy was compared to the monotherapy. To control for the overall type I error rate, a type I error rate of 0.025 was planned for the comparison of each combination treatment with monotherapy. The analytic approach for the two comparisons was identical. A chi-square test was used to compare the remission rates across the treatment groups. Fisher’s exact test was used when the expected cell frequencies were less than 5. For binary outcomes (e.g., remission), bivariate logistic regression models were fit to estimate the effect of treatment on outcome. Multivariable logistic regression models were then fit to control for the effect of regional center and baseline characteristics that were not balanced across treatment groups. A similar approach was used for discrete outcomes with more than two levels, except a polytomous logistic regression model was used. For continuous outcomes, a t test was used to compare the means when distributions were normal, and the nonparametric Kruskal-Wallis test was used when distributions were nonnormal. Linear regression models were used to compare the means after controlling for regional center and baseline characteristics not balanced across treatment groups. A general linear model with a negative binomial distribution and log link was estimated for outcomes with severely nonnormal distributions (the last number of worsening adverse events indicated by the Systematic Assessment for Treatment Emergent Effects and the score on the IDS-C anxiety subscale).
Discussion
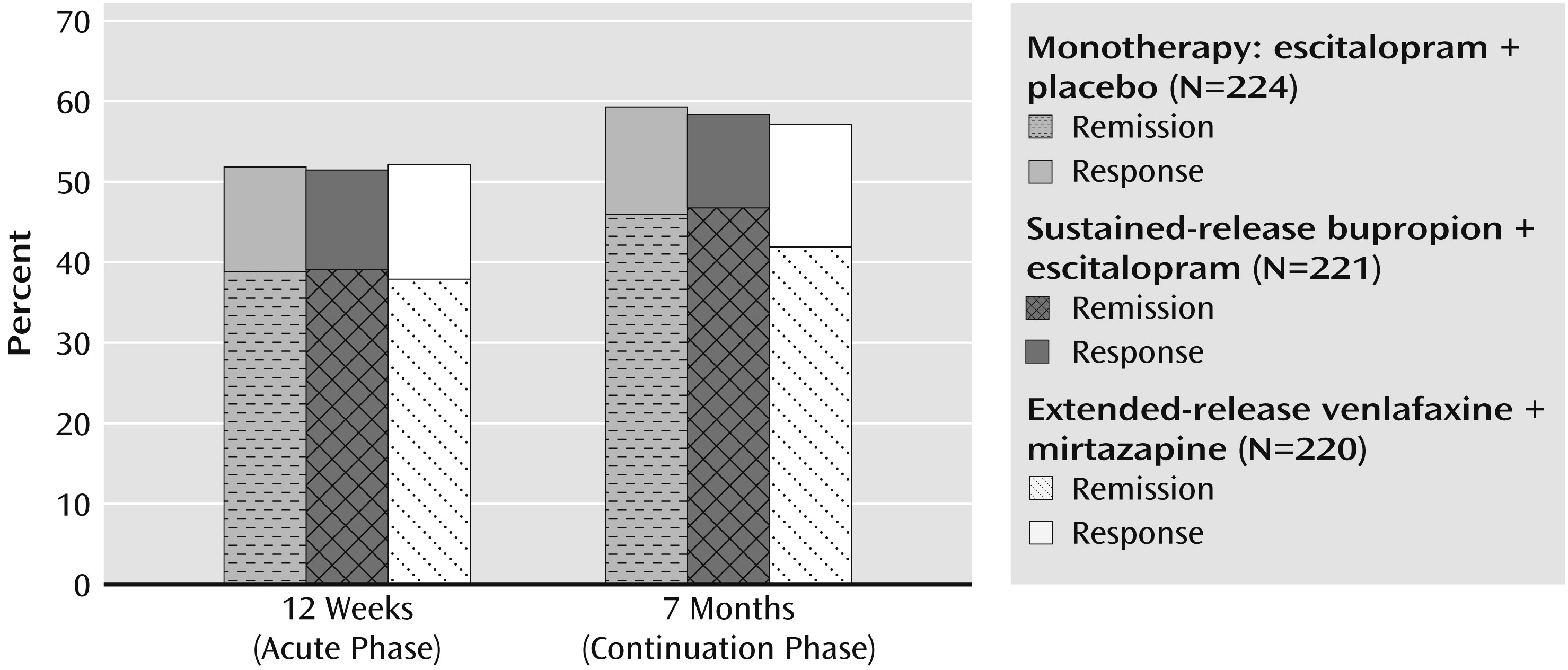
The study has four key findings: 1) remission and response rates were not different at 12 weeks, 2) remission and response rates were not different at 7 months, 3) the effects of the three treatments on quality of life and on work and social adjustment were not different, and 4) extended-release venlafaxine plus mirtazapine was associated with a greater side effect burden at 12 weeks and 7 months than escitalopram plus placebo and a higher number of worsening adverse events than escitalopram plus placebo at 7 months. We found no clinical advantage over escitalopram-placebo from either combination of antidepressant medications in terms of either remission or response rates at either 12 weeks or 7 months. The remission rates approximated those expected on the basis of monotherapy studies of chronic depression (
5,
6). Both combination treatments had more side effects (in terms of frequency, intensity, or burden) than escitalopram-placebo in both the acute and continuation phases. Attrition rates, however, were not different across the three treatment groups in either phase of treatment. The venlafaxine-mirtazapine group was at particularly greater risk for side effects. In fact, it had significantly greater worsening of side effects than escitalopram-placebo despite the fact that the mirtazapine dose was not high (about 20 mg/day).
Prior reports have suggested that the response to either medication combination would exceed the effects of monotherapy. An open study of 49 patients (
41) given escitalopram (up to 40 mg/day) plus sustained-release bupropion (400–450 mg/day) found a 63% remission rate at week 8. Blier et al. (
18) compared mirtazapine, paroxetine, and the combination in a 6-week double-blind, randomized, controlled trial conducted at two research clinics with clinically referred patients and symptomatic volunteers (N=61). Remission rates at 6 weeks were 19% for mirtazapine, 26% for paroxetine, and 43% for the combination. Most of these patients had melancholic symptom features and either had nonrecurrent depression or had an index episode lasting less than 1 year. Drug doses included up to 45 mg/day of mirtazapine and paroxetine amounts that could exceed 30 mg/day (average final or exit doses not reported).
In a recent, larger 6-week double-blind acute randomized, controlled trial, Blier et al. (
13) compared fluoxetine (20 mg/day) with mirtazapine (30 mg/day) in combination with fluoxetine (20 mg/day), extended-release venlafaxine (225 mg/day), or sustained-release bupropion (150 mg/day). Each combination had a remission rate (46%–58%) that exceeded that of fluoxetine alone (25%). In the study, 76% of the participants met melancholia criteria, 63% had recurrent major depression, and 61% had an index episode longer than 1 year. It is interesting that the response rates were not significantly different (54% for fluoxetine, 68% for mirtazapine-fluoxetine, 73% for mirtazapine-venlafaxine, 65% for mirtazapine-bupropion). Of note, this 6-week study may have been too brief to allow the full benefit of fluoxetine to be expressed (
42).
There are several other possible explanations for why our findings differ from those of Blier et al. (
13). The studies differ in terms of length of treatment, primary outcome, and scales used to assess outcomes. Our results are not accounted for by either differential attrition across the three treatments or baseline differences. On the other hand, our participants differed from those studied by Blier et al. Neither participant group was treatment resistant. However, participants in our study were required to have chronic and/or recurrent depression. In fact, there were far more chronically ill participants in our study than in the one by Blier et al. In addition, 62%–85% of their participants had melancholic features (compared to only 20% in this study). Some reports (
17,
43) suggest better efficacy, i.e., drug-placebo differences, for inpatients (who are more likely to have melancholic features) with combination medications or dual-action medications. In addition, a meta-analysis by Perry (
44) revealed that broader-action agents (e.g., tricyclic antidepressants) have far greater efficacy than SSRIs in melancholic depression.
To evaluate the potential impact of chronicity on outcome, we reanalyzed the data. Chronicity was associated with lower remission rates across all three treatment cells. Specifically, in the bupropion-escitalopram group, 37.2% of the patients with episodes of chronic depression had remissions, whereas the remission rate was 41.0% for non-chronic depression. Analogous rates were 35.5% versus 43.1% for escitalopram-placebo and 34.9% versus 41.9% for venlafaxine-mirtazapine. We conducted a similar analysis to compare patients with and without melancholic features from the current study. Melancholic features were associated with more axis I comorbidity, greater symptom severity, and more suicidal plans and thoughts. However, remission rates ranged from 30.0% to 39.1% for those with melancholic features and from 37.5% to 39.5% for those without. There were no differences across medication groups. Thus, neither the difference in the proportion with chronic illness nor the difference in melancholia seems to account for why our results differ from those of Blier et al. (
13). While we enrolled the kinds of patients (i.e., those with chronic or recurrent depression) for whom most clinicians would be likely to consider antidepressant medication combinations (
45), we cannot rule out potential impact on outcomes from one or more unknown baseline features.
This group with chronic and/or recurrent depression had high rates of self-reported emotional, sexual, or physical abuse before age 18, and a high proportion had anxious features. While these features could have reduced the overall benefit of any one treatment, they would be unlikely to obscure differences between treatment cells, given their proportional distribution across the cells.
Perhaps the differences between the present study and the results reported by Blier et al. (
13) are due to the specific antidepressant medications and doses that we used and to the doses that were administered. There is evidence for the efficacy of venlafaxine plus mirtazapine (
13,
35) and bupropion plus escitalopram (
16,
34). Carpenter et al. (
46) found that mirtazapine, when used as an adjunct to previously ineffective SSRIs alone, was more effective than placebo in treating depression. In fact, Blier et al. (
13) used the venlafaxine-mirtazapine combination and Stewart et al. (
41) used bupropion plus escitalopram in their trials, although the doses were higher in both of them. The rationale for a higher venlafaxine dose is that the effect on the norepi-nephrine system (
17,
47) is only realized at doses of at least 225 mg/day. Mirtazapine has antagonistic effects that are modest at 15 mg/day and more clearly evident at 30 mg/day. Thus, as suggested by Blier (personal communication), the doses in CO-MED may have been insufficient in a large enough proportion of participants to preclude the benefit otherwise available from the combination. In an attempt to evaluate that notion, we identified the 86 participants who reached 225 mg/day of venlafaxine and 30 mg/day of mirtazapine at any time during treatment. Their remission rate was 33.7% at 12 weeks and 41.9% at 7 months. These results do not suggest that underdosing was the cause of the poor performance of this combination.
It remains an unanswered question whether these larger doses (if they are required to achieve an advantage for antidepressant combinations) are achievable in practice with more representative patients who have chronic and/or recurrent major depressive disorder and more concurrent axis I and III disorders.
This study had several limitations. While larger than many studies, the study group may not be representative of the universe of outpatients with chronic and/or recurrent major depression. As noted, the doses used may not have been sufficient to realize the full potential value of combination antidepressant medications. The results for the continuation treatment phase are limited by the fact that the subjects were not rerandomized or stratified by level of improvement following the acute phase. Finally, the clinicians were not blind to treatment, and a structured interview was not used to establish axis I diagnoses.
In summary, in outpatients with chronic and/or recurrent major depressive disorder, there appears to be no advantage to either medication combination over escitalopram alone as a first-step treatment for nonresistant depression. Some combinations may incur a risk of higher side effect burden. This conclusion is conditioned on the doses employed.