2. Demographics and Clinical Course
Anti-NMDAR encephalitis was originally identified linking a syndrome with prominent psychiatric manifestations in the context of encephalitis in four young women with ovarian teratoma (
Vitaliani et al., 2005). It is now appreciated that children and males can be affected and that the same neurologic syndrome may develop either without a tumor or as a paraneoplastic manifestation of an underlying teratoma (
Dalmau et al., 2008;
Viaccoz et al., 2014). The frequency of an underlying teratoma, and very rarely other tumors, is dependent on patients' sex and age (
Florance et al., 2009). Children under 12 years of age and male patients rarely have a tumor (
Titulaer et al., 2013). Regardless, this disorder overwhelmingly affects young women, though patients as young as 2 months and as old as 85 years have been reported (
Titulaer et al., 2013;
Armangue et al., 2014). The clinical course begins in most instances with a viral prodrome, followed by prominent psychiatric symptoms such as psychosis (delusional thinking, hallucinations), agitation, and confusion (
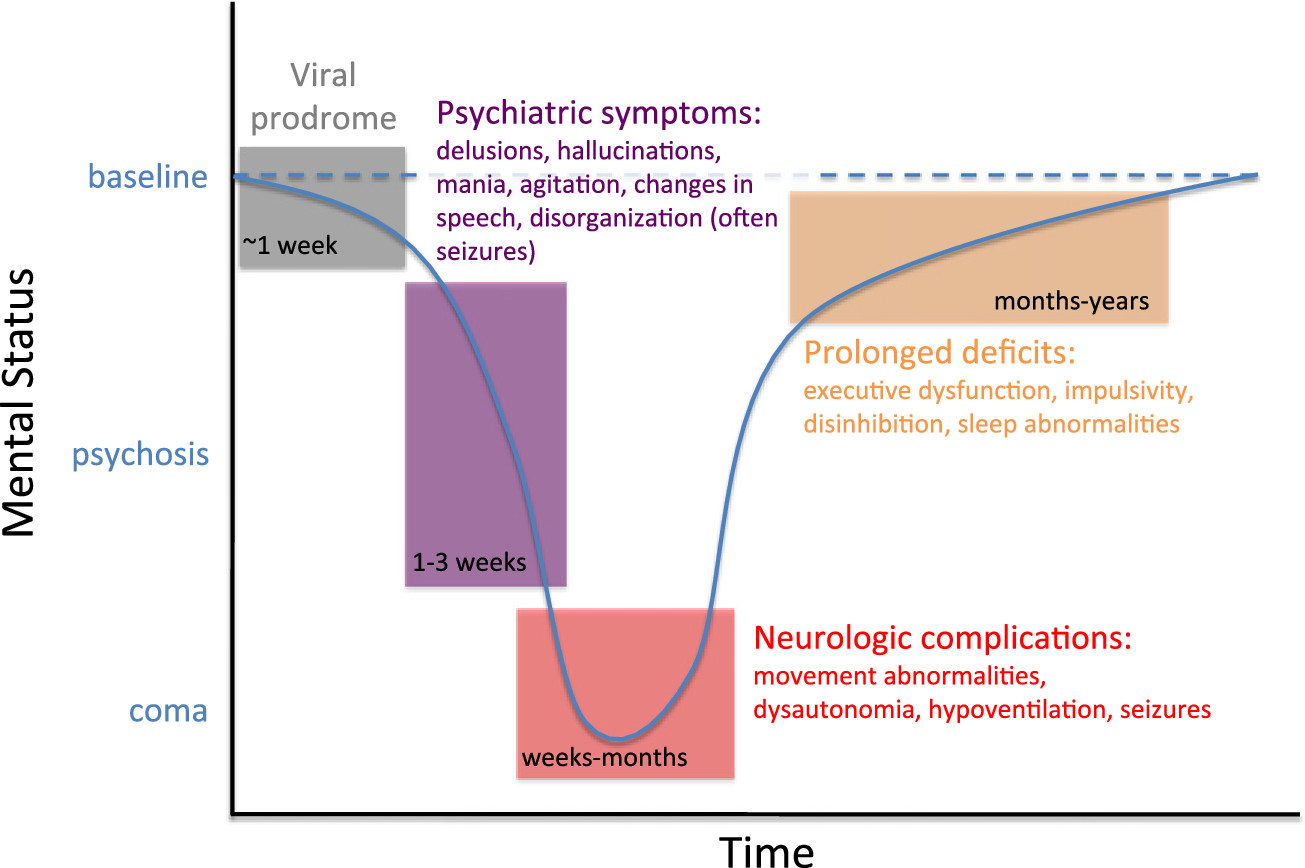
Fig. 1) (
Dalmau et al., 2008;
Kayser and Dalmau, 2011). Most cases progress to include severe neurological features like seizures, movement abnormalities, autonomic instability, or hypoventilation, often requiring ICU-level care (
Dalmau et al., 2008;
Irani et al., 2010;
Titulaer et al., 2013). Recent work has demonstrated that early and aggressive immunosuppression along with removal of tumor (if present) lead to positive outcomes, with 80% of patients returning to near baseline level of function (Titulaer et al., 2013;
Viaccoz et al., 2014). Relapse is less common than once thought, particularly with effective management, as only ∼ 10–15% of patients relapse in a 2 year period (
Titulaer et al., 2013). Overall, this once often-fatal encephalitis is now clinically recognizable, diagnosable by presence of antibodies in CSF, and treatable with immunosuppression, all due to a detailed understanding of the underlying disease process.
3. Mechanisms of Disease
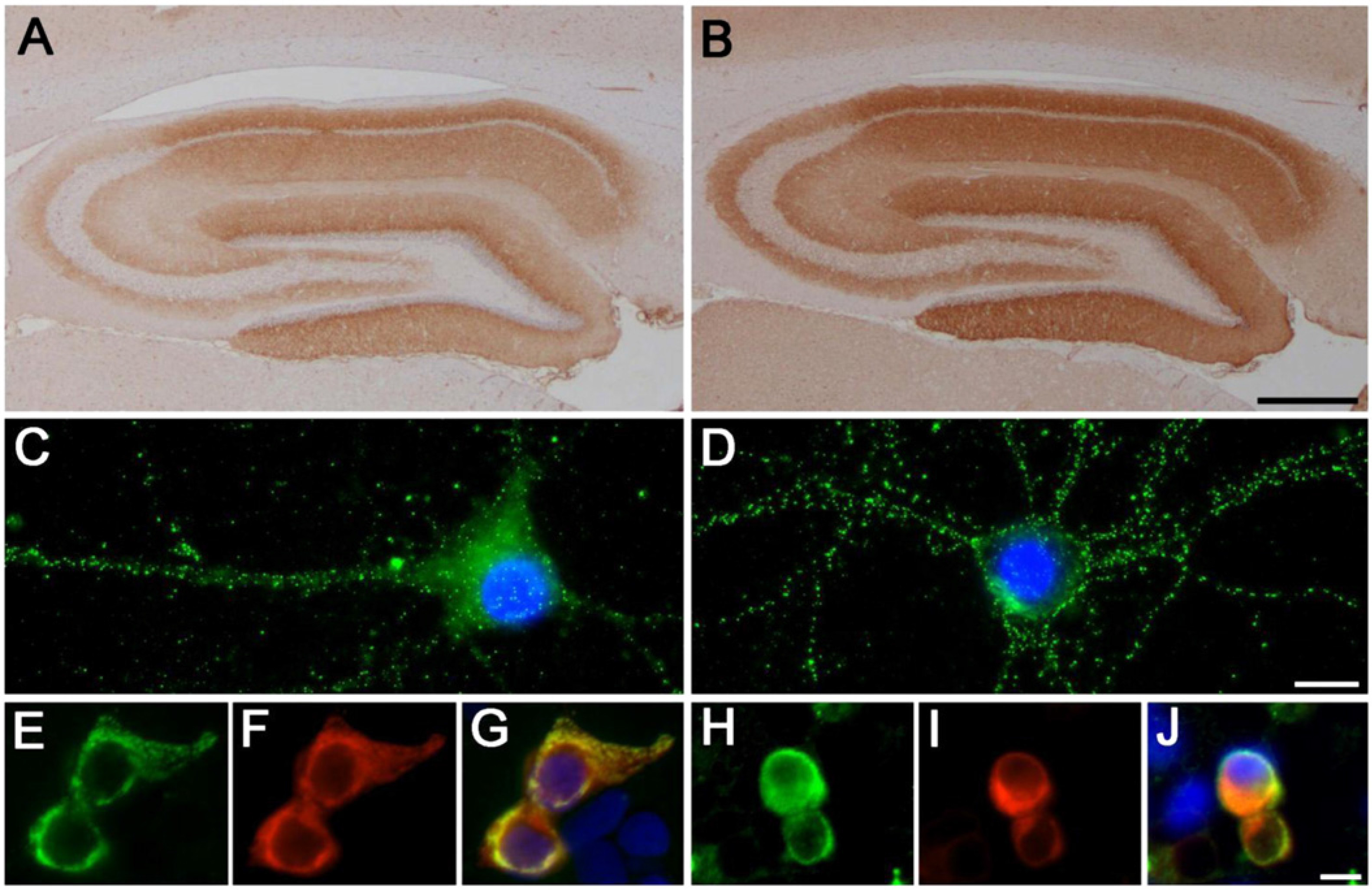
The cellular mechanisms underlying anti-NMDAR encephalitis are increasingly well understood. Binding of IgG antibodies to NMDARs induces a reversible internalization of the receptors from both synaptic and extrasynaptic space, with relative sparing of other glutamate receptors and excitatory scaffolding proteins; the number of synapses remains unchanged, and dendritic structure and cell survival is not perturbed (
Hughes et al., 2010). Internalization of NMDARS occurs through antibody binding, capping, and cross-linking of the receptors, and loss of NMDARs from the cell surface correlates with antibody titer (
Hughes et al., 2010). Notably, CSF antibody titers correlate better than serum titers with clinical outcome and relapses (
Gresa-Arribas et al., 2014). The removal of NMDARs from the cell surface is supported not only by immunohistochemical evidence in vitro and in vivo, but also electrophysiological measures, indicating a decrease of synaptic NMDAR-mediated currents due to the low level of receptors induced by auto-antibodies rather than a direct antibody blockade (
Hughes et al., 2010;
Moscato et al., 2014). Decreased synaptic NMDAR content also leads to reduced synaptic plasticity, as treatment of rodent neurons with patient CSF blocks molecular signatures of long-term potentiation (LTP) (
Mikasova et al., 2012). The effect on NMDARs does not appear to be specific to excitatory neurons, as NMDARs on inhibitory neurons are similarly internalized (
Moscato et al., 2014). Interestingly, in vitro investigations into the time frame of these cellular phenomena show that in neurons continuously exposed to patients' CSF antibodies, the process of NMDAR internalization becomes microscopically visible in 2 h, reaching the lowest level of NMDAR receptor density after 12 h; subsequently there is a steady state of low levels of synaptic NMDAR for as long as the neurons are exposed to patients' antibodies (
Moscato et al., 2014). These effects were independent of receptor activity. Moreover, while patient NMDAR auto-antibodies did not induce compensatory changes in glutamate receptor gene expression, they caused a decrease in inhibitory synapse density onto excitatory hippocampal neurons (
Moscato et al., 2014). These findings suggest a potential mechanism of increased excitability, which is in line with the in vivo effects (increase of extracellular glutamate) of patients' antibodies after injection in the premotor cortex of rats (
Manto et al., 2010).
Does exposure to antibodies in animal models result in similar behavioral effects as in humans? Recent work has found that prolonged cerebroventricular infusion of patient antibodies into mice results in progressive memory deficits, anhedonia, and depressive-like behaviors (
Planaguma et al., in press). These behavioral changes correlated with detection of a progressive (over days) increasing concentration of brain-bound NMDAR antibodies and parallel decrease of the density of synaptic and extrasynaptic NMDAR clusters. After the infusion of antibodies stopped there was progressive (over days) clinical improvement associated with restoration of NMDAR levels, a feature strikingly conserved from in vitro work to the clinical experience of patients. In sum, multiple lines of evidence now demonstrate that anti-NMDAR antibodies in this disorder cause reversible internalization of NMDARs from the synapse in humans and rodent models, resulting in behavioral manifestations across species.
4. Anti-NMDAR Antibodies and Psychosis
The prominence of psychotic symptoms in anti-NMDAR encephalitis has been of tremendous interest to the psychiatric community by raising the possibility of an identifiable, treatable subtype of psychosis. This is unlikely to be the case if prominent neurological symptoms always accompany psychiatric manifestations in the presence of CSF auto-antibodies. Do patients with known anti-NMDAR encephalitis experience isolated psychiatric symptoms either at initial presentation or even in relapse? Examination of nearly 600 cases (
Kayser et al., 2013) has shown that, while rare, some patients may demonstrate only psychiatric symptoms without any neurological involvement during the first disease episode or in a relapse episode.
Fig. 2 demonstrates NMDAR antibodies in a single patient at two different time points: during the recovery phase of the initial presentation (severe neurological involvement with ICU-level care and intubation) and during a relapse episode two years later (pure psychiatric symptoms with prominent psychosis and aggression). Remarkably, NMDAR titers were higher in relapse (1:80 dilution with continued reactivity) than during the recovery phase of initial presentation (1:5 dilution). Prolonged time to treatment in some cases with isolated psychiatric symptoms confirms that isolated psychiatric episodes are truly monosymptomatic and not simply a result of early intervention that spared clinical worsening (
Kayser et al., 2013). These patients do not differ with regard to demographics, presence or absence of tumor, psychiatric symptoms constellation, or outcomes from the population of anti-NMDAR encephalitis as a whole (
Kayser et al., 2013). It seems possible, therefore, that patients with anti-NMDAR encephalitis defined by IgG subtype antibodies reacting with the GluN1 subunit can manifest with psychiatric symptoms in isolation. Notably, symptoms in all of these patients responded to immunomodulatory therapy (
Kayser et al., 2013).
Numerous groups have also screened patients diagnosed with primary psychiatric disorders to determine whether a subset of schizophrenia patients, for example, actually harbor anti-NMDAR auto-antibodies and might respond to immunosuppression. Importantly, anti-NMDAR encephalitis and the associated psychotic symptoms are caused specifically by IgG antibodies recognizing the GluN1 subunit of the NMDAR (
Dalmau et al., 2008). The clinical significance of IgG antibodies that target other NMDAR subunits or of IgA or IgM subtypes recognizing NMDARs is unknown, though connections have been made between these subtypes and dementia and viral encephalitis (
Pruss et al., 2012a,
b;
Doss et al., in press). Examination of serum obtained at symptom onset from 80 patients with new-onset psychosis who 1 year later met criteria for schizophrenia-spectrum illness revealed no presence of IgG GluN1 antibodies in either patients or controls (
Masdeu et al., 2012). These results are consistent with another smaller study that failed to demonstrate GluN1 IgG auto-antibodies in patients with schizophrenia (
Rhoads et al., 2011). Haussleiter and colleagues also examined 50 psychosis patients including some with chronic psychosis (nearly ¾ of all patients had symptoms for at least 5 years) and found no evidence of anti-NMDAR antibodies (
Haussleiter et al., 2012). Other groups have reported presence of serum NMDAR auto-antibodies in a subset of patients with psychosis, but it does not appear that these are IgG GluN1 antibodies known to cause schizophrenia-like symptoms associated with anti-NMDAR encephalitis.
Zandi et al. (2011) found IgG antibodies recognizing GluN1/GluN2 NMDAR subunits in serum from 3 of 46 patients with early onset schizophrenia; no patients with chronic schizophrenia harbored such antibodies. The specific NMDAR subunit targeted was not determined (GluN1 vs GluN2) and other control groups or additional confirmatory tests were not included. Indeed, using the same assay, these authors recently reported serum NMDAR antibodies in 23% of patients who had disorders considered unlikely to be immune mediated (e.g., posterior fossa glioma, atypical motor neuron disease, leukodystrophy, maple syrup urine disease, or idiopathic pure cerebellar syndrome) (
Zandi et al., 2014). Given these findings, Zandi et al., have modified the interpretation of their assay scoring system, so that at least one of the three schizophrenia patients initially reported to harbor pathogenic auto-antibodies would currently be considered unlikely to have an immune associated disorder.
In a large study,
Steiner et al. (2013) examined serum from 459 patients with either schizophrenia, major depression, or borderline personality disorder. Only two patients with classic anti-NMDAR encephalitis that had been misdiagnosed were found to harbor GluN1 IgG antibodies, emphasizing the importance of familiarity with this syndrome among psychiatrists. Of note, ∼ 10% of patients with schizophrenia were found to have IgA and/or IgM subtype antibodies reacting with GluN1/GluN2 subunits (not GluN1 alone) (
Steiner et al., 2013). This work initially suggested that NMDAR autoimmunity could be of relevance in schizophrenia, but that the antibody subtypes and targets are distinct from that in anti-NMDAR encephalitis. However, recent work by the same authors re-examining the sera of the control group showed that the frequency of IgA and IgM antibodies was in fact similar to that of the patients (
Steiner et al., 2014). These findings are in line with the study of Hammer et al. who reported a high percentage (∼ 10%) of NMDAR autoantibody positivity with almost entirely IgA and IgM subtypes in both patients and controls (
Hammer et al., 2013). Overall, these studies highlight the fact that in contrast to IgG antibodies to GluN1 which are highly specific of anti-NMDAR encephalitis (
Gresa-Arribas et al., 2014), serum IgA and IgM antibodies to NMDAR subunits are similarly elevated in healthy controls and many disease groups examined, and that the clinical significance of IgA and IgM antibodies is uncertain (
Titulaer and Dalmau, 2014).
All patients with anti-NMDAR encephalitis have CSF IgG antibodies against GluN1, almost always accompanied by intrathecal antibody synthesis (
Dalmau et al., 2008). Given that IgA and IgM NMDAR antibodies in serum lack disease specificity and the occurrence in healthy subsets questions their clinical significance (
Busse et al., 2014), it has been suggested that these antibodies might increase symptom severity in schizophrenic patients with history of neurotrauma and blood–brain-barrier disruption (BBB) (
Hammer et al., 2013). However, none of the studies proposing these theories demonstrated relevant neurotrauma or BBB dysfunction, and unfortunately, none of them included CSF investigations. More recently the same authors examined serum of 1703 healthy individuals and 2533 patients with neuropsychiatric illness (schizophrenia, affective disorders, Parkinson, ALS, personality disorder), finding that 10% of all disease groups and normal subjects had NMDAR antibodies (mostly IgA and IgM) (
Dahm et al., 2014). Patients with anti-NMDAR encephalitis were not included in any of these studies; yet, the authors concluded that serum antibodies of any immunoglobulin isotype against the GluN1 subunit of the NMDAR are not a disease indicator (
Ehrenreich and Steiner, 2014).
5. Convergent Mechanisms of Anti-NMDAR Encephalitis and Schizophrenia
The glutamate hypothesis of schizophrenia is founded largely on the psychotic symptoms associated with NMDAR antagonists, such as PCP and ketamine (
Javitt, 2012;
Javitt et al., 2012;
Moghaddam and Krystal, 2012). Many lines of evidence, of which anti-NMDAR encephalitis is one of the most recent, have come to support the view that reduced NMDAR synaptic content might play a significant role in psychotic symptoms. Anti-NMDAR encephalitis demonstrates clearly that NMDAR hypofunction can result not only in psychosis, but also in cognitive compromise, mood changes, and other symptoms of schizophrenia. Targeted, inducible, genetic approaches in rodent models have now revealed that dysfunction of NMDARs in cortical GABAergic interneurons is particularly relevant for schizophrenia-like processes (
Belforte et al., 2010). Notably, anti-NMDAR encephalitis produces a similar constellation of symptoms, although the antibody reactivity is not limited to that subset of neurons, but found in the entire brain and predominantly regions with high neuronal and NMDAR density, such the hippocampus (
Moscato et al., 2014). Interestingly, imaging-based approaches in humans have directly implicated glutamatergic dysfunction leading to connectivity and/or functional changes in hippocampus (
Schobel et al., 2013) and cortex (
Driesen et al., 2013). Disruption of NMDAR function in discrete brain regions may therefore give rise to similar schizophrenia-like phenotypes.
How might anti-NMDAR encephalitis inform our understanding of schizophrenia at large? Studies of NMDAR-modulating drugs have proven incredibly powerful over the past 25 years (
Javitt, 2012), while leveraging NMDAR auto-antibodies to study psychosis has just begun; there are many potential roads forward. First, there is more to learn regarding how these antibodies demonstrate regional specificity in the brain, whether related to proximity to the ventricles, density of receptors, or some other type of susceptibility in hippocampus. Second, functional imaging will provide important insight on the similarities between the symptoms occurring with NMDA receptor antagonists and the disorder mediated by NMDA receptor antibodies. There is preliminary evidence that the alterations of connectivity using resting state functional MRI have notable parallels with those obtained with NMDAR antagonists (
Finke et al., 2013). Finally, the link between infection and schizophrenia is already well-appreciated, and suggests involvement of immunologic processes (
Benros et al., 2011). In fact, even prior to discovery of NMDAR encephalitis, it was hypothesized the schizophrenia is a mild encephalitis (
Bechter, 2013); many patients exhibit symptoms beyond psychosis (cognitive impairment, catatonia, autonomic dysfunction) though pathognomonic auto-antibodies remain elusive. NMDAR encephalitis is often preceded by prodromal symptoms suggesting a viral process, and there is recent evidence that herpes simplex encephalitis can trigger NMDAR and other types of synaptic autoimmunity (
Prüss et al., 2012a;
Armangue et al., 2014). The link between herpes simplex encephalitis and NMDAR autoimmunity only applies to a very small subset of patients with anti-NMDAR encephalitis, but it is robust and represents a proof of concept that a viral process can develop into an immune-mediated dysfunction of synaptic transmission. Thus this syndrome might provide insights into how infectious etiologies are associated with subsequent psychosis.