Prognosis and Improved Outcomes in Major Depression: A Review
Abstract
Depression: a major and relentless burden
Prognostic variables for treatment outcomes in MDD
Clinical Variables
| Marker | Outcome | References |
|---|---|---|
| Clinical | ||
| Short duration of untreated disease | ↑ | 5–10 |
| Early response to treatment | ↑ | 9, 12–15 |
| Lower baseline function | ↓ | 21, 22, 24 |
| Psychiatric comorbidity (anxiety disorders, PTSD, OCD, personality, cumulative) | ↓ | 26, 40–46 |
| Physical comorbidity (pain, cardiovascular, neurological, cumulative) | ↓ | 47, 48, 50–56 |
| Stressful life events, childhood maltreatment | ↓ | 33–37 |
| Treatment resistance | ↓ | 28, 109 |
| Neuroimaging | ||
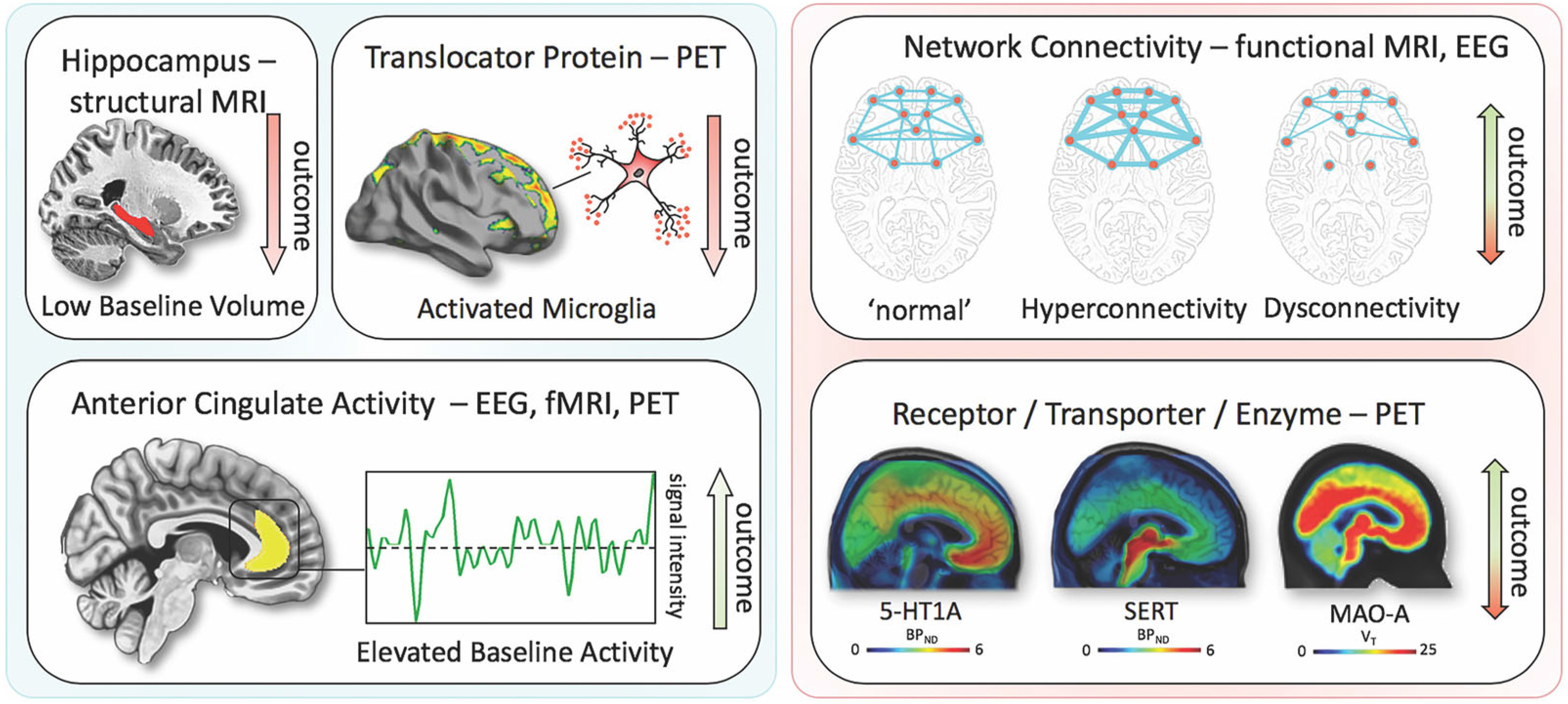
| Low baseline hippocampal volume—sMRI | ↓ | 59, 60 |
| High baseline activity in the anterior cingulate cortex– fMRI, EEG, PET | ↑ | 60, 70, 71 |
| Microglial activation (TSPO-PET) | ↓ | 80–82 |
| rsfMRI in pathophysiologic regions | ↓↑ | 69 |
| Key proteins of the serotonergic system (MAO-A, SERT, 5-HT1A) | ↓↑ | 72–77 |
| Blood | ||
| Plasma BDNF increases in response to treatment | ↑ | 93 |
| IL-6 decreases during treatment | ↑ | 83 |
| High TNFα levels after treatment | ↓ | 86 |
| High baseline CRP levels | ↓ | 84, 85 |
| Candidate genesa | ||
| BDNF—Val66Met Met allele in Asians | ↑ | 203 |
| SLC6A4–5-HTTLPR, l-Allele | ↑ | 204 |
Psychosocial Variables
Environmental Stress and Stressful Life Events (SLEs)
Psychiatric and Physical Comorbidities
Neuroimaging Markers of Treatment Outcomes
Blood-Based Markers of Disease Outcomes
Outcome and Genetic and Epigenetic Links
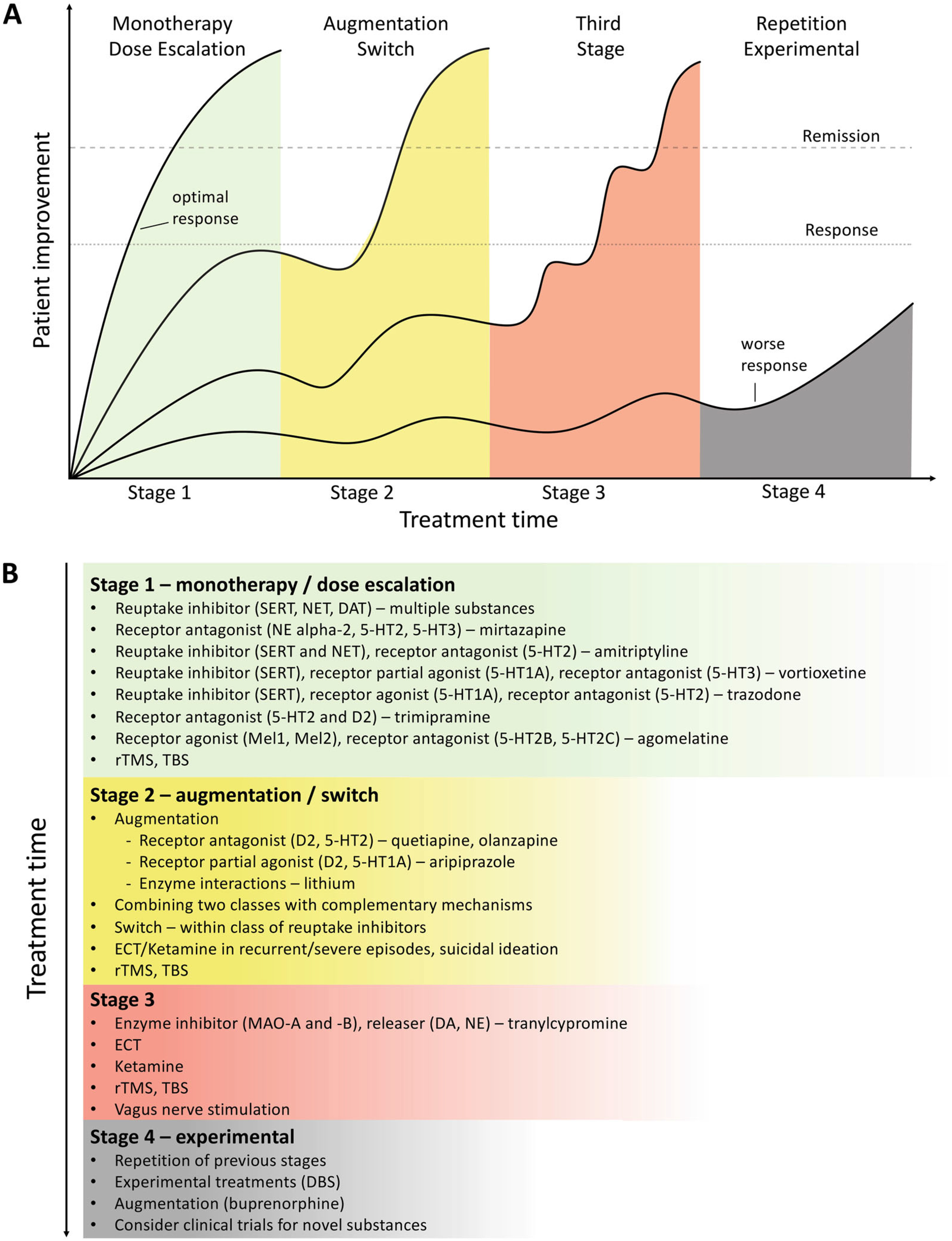
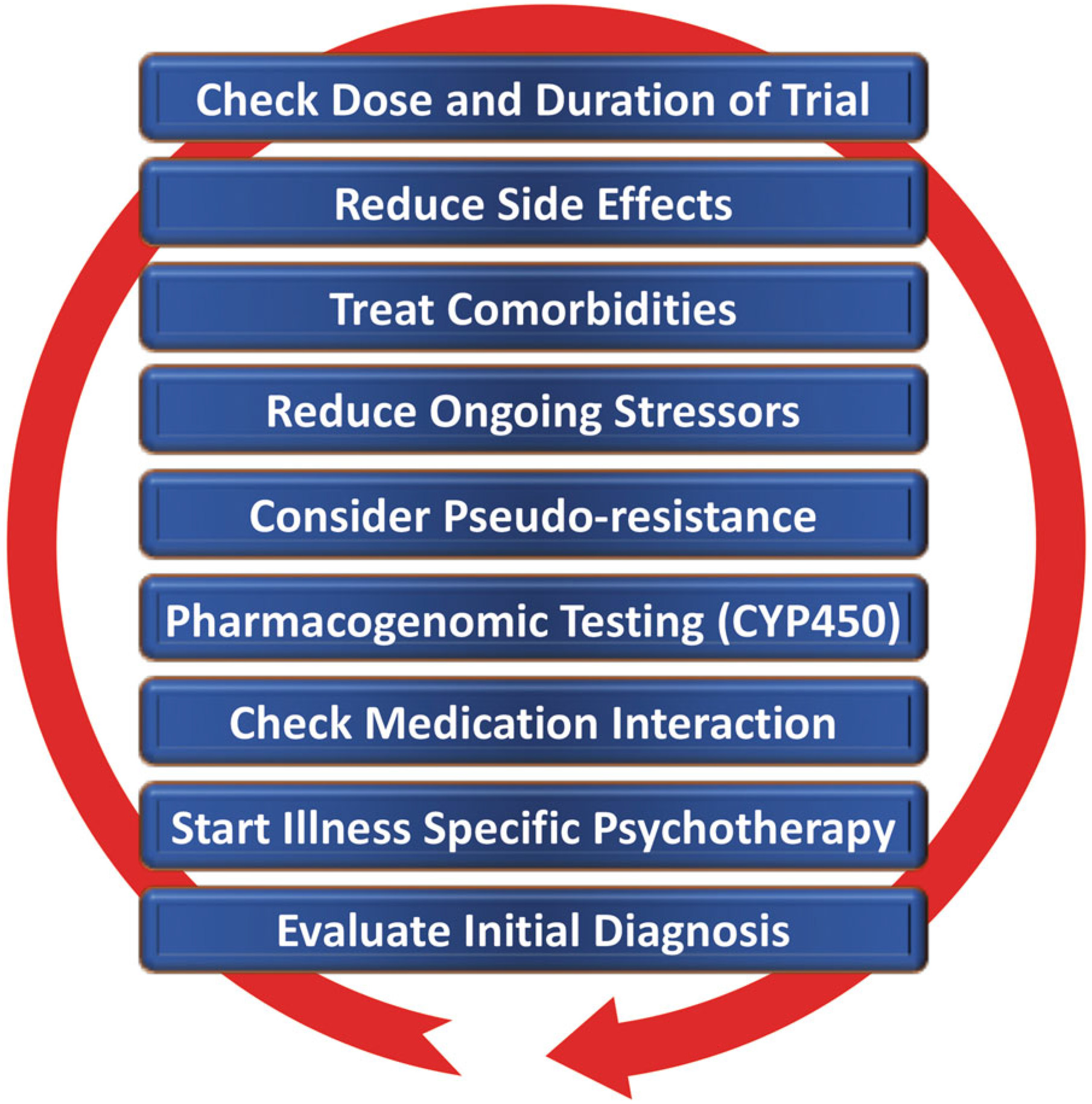
Clinical Subgroups, TRD, and Treatment Outcomes
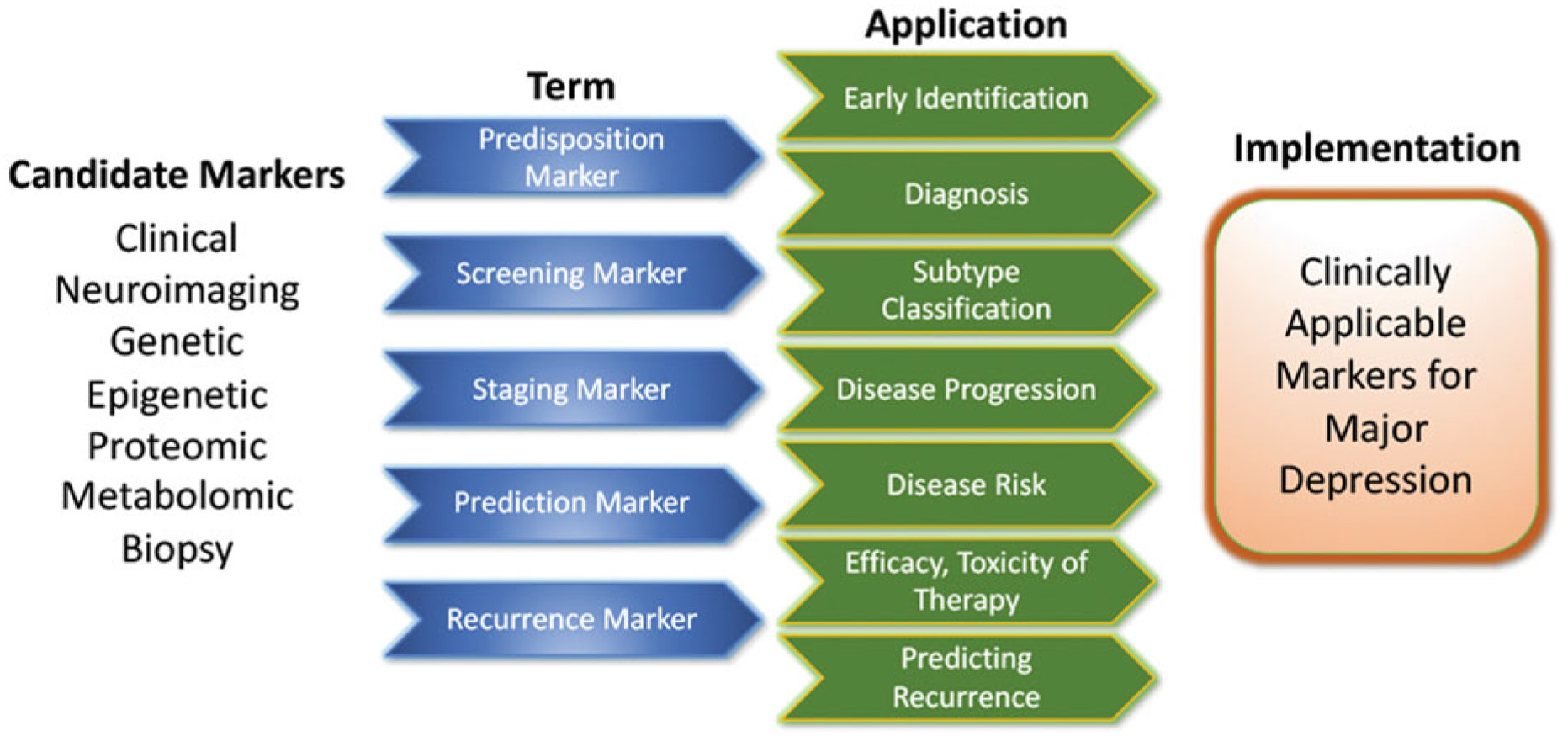
Novel and existing strategies to improve treatment outcomes
Early Identification, Prevention, and Early Treatment
Modeling Environmental Impact on Predisposition
Developing Markers for Subgroup Identification and Disease Course
Algorithm- and Guideline-Based Treatments
| Name/Organization | URLa/reference | Country, Year |
|---|---|---|
| World Federation of Societies of Biological Psychiatry (WFSBP) consensus papers and treatment guidelines | www.wfsbp.org | Worldwide, 2015, 2013, 2007 |
| American Psychiatric Association Practice Guidelines (APA) | www.psychiatryonline.org/guidelines | USA, 2010 |
| British Association for Psychopharmacology | www.bap.org.uk/guidelines | UK, 2015 |
| Canadian Network for Mood and Anxiety Treatments (CANMAT) | www.canmat.org | Canada, 2016 |
| Institute for Clinical Systems Improvement (ICSI) Healthcare Guideline for Major Depression in Adults in Primary Care | www.icsi.org | USA, 2016 |
| S3 Guidelines | www.leitlinien.de/nvl/depression | Germany, 2017 |
| Therapy resistant depression guideline | www.oegpb.at | Austria, 2017 |
Reducing Placebo Response in Clinical Trials While Harnessing Placebo Effects in Clinical Treatment
Novel Antidepressant Treatments
Ketamine.
Esketamine hydrochloride.
Other rapid acting and novel antidepressants.
Transcranial Stimulation Paradigms
Electroconvulsive Therapy (ECT)
Magnetic Seizure Therapy (MST)
Vagus Nerve Stimulation (VNS)
Deep-Brain Stimulation (DBS)
Conclusions
| • Enormous improved outcomes are needed in MDD |
|---|
| • Candidate clinical, neuroimaging, blood, and genetic markers exist but need to be improved to be applicable for routine clinical care |
| • Early identification and treatment facilitate better outcomes |
| • The advantages of existing treatments may be harnessed by standardized sequential use |
| • Novel antidepressants—some with rapid-acting mechanisms—have high potential for approval |
| • Brain stimulation techniques such as TMS, TBS, ECT, and DBS are evolving and are an important, often underused treatment option |
| • Treatment strategies for chronic patients exist, but more research needs to focus on “third-line-and-beyond” therapeutics |
REFERENCES
Information & Authors
Information
Published In


History
Authors
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).