Parkinson’s disease (PD) is a common neurological disorder in older adults, which typically presents with tremor and impairment of movement initiation. Patients with PD are up to six times more likely to develop dementia than the general population, and a specific syndrome of PD dementia (PDD) has been defined (
1). Up to 70% of patients with PD ultimately develop PDD, on average 10 years after their PD diagnosis (
1).
Nonmotor symptoms of PD, including depression and psychosis, are common (
2). Cross-sectional studies have shown that nearly 30% of patients with PD and 50% of patients with PDD develop visual hallucinations (VHs) during their illness; these are often of people or animals and can affect patients’ quality of life (
3). The phenomenology is very similar to that of hallucinations in dementia with Lewy bodies (DLB) (
1). It is now thought that there is a continuum of visual symptoms in PD from minor visual phenomena to formed VHs. Several theories have been suggested to explain VHs in PD, but currently the evidence suggests that visual hallucinations in PD are a result of changes in cortical visual processing rather than an ocular deficit (
4). While hallucinations do occur in other modalities (e.g., auditory), these are less prevalent than VHs (
3).
Neuroimaging has been used in several studies to investigate VHs in PD. Two small (N=21 and N=19) [
18F]-fluorodeoxyglucose positron emission tomography (FDG-PET) studies of individuals with PD and mild cognitive impairment found that those with VHs had reduced bilateral occipital and parietal uptake (
5,
6). Because the fluorodeoxyglucose is distributed intravascularly according to blood flow and then taken up by cells but not further metabolized, the reduced occipitoparietal uptake largely reflects diminished cerebral perfusion. Another small (N=28) FDG-PET study demonstrated reduced uptake in temporal and parietal brain regions involved in the ventral visual processing stream (
7). A study of eight nondemented PD patients with and 11 without VHs found evidence of increased frontal uptake only in those with VHs (
8). A similar sized single-photon emission computerized tomography (SPECT) study (N=24 with VHs and N=41 without) found no evidence of occipital lobe hypoperfusion, but some evidence of temporal lobe hypoperfusion in those with VHs (
4). A small functional MRI (fMRI) study (N=24) of brain activation during visual tasks found that participants with PD plus VHs had an unusual pattern of visual pathway activation. Posterior regions of the brain (occipital, parietal, and temporal) were activated less and frontal visual processing areas activated more during visual tasks in those with VHs compared with those without VHs (
9). A case report from the same group with fMRI data obtained during VHs again showed increased activation of anterior brain regions and relative deactivation of posterior brain regions during VHs (
10). The authors suggested that aberrant top-down processing may lead to VHs. A modestly sized fMRI study (N=38) by Yao et al. (
11) lent further support to the top-down model of VHs in PD.
It has been shown in postmortem tissue that α-synuclein burden in the amygdala, frontal, and temporal regions was associated with VHs, but only in individuals who had dementia (
12). A later, much larger study in which the mean Mini-Mental State Examination score of brain donors had been ≤20 within 1 year of death found again that Lewy body burden in multiple brain regions was associated with VHs. The authors also found that there was coexistent Alzheimer’s pathology (plaques and tangles) in many of the same brain areas and suggested that both Alzheimer’s disease and PD pathology contributed toward VHs (
13).
Cholinergic failure in PD is thought to contribute to the associated neurocognitive deficits. Neuronal loss from the basal forebrain cholinergic nuclei (including the nucleus basalis of Meynert [nbM] from which cortical cholinergic innervation originates and nucleus of the diagonal band of Broca) (
14) and brain stem cholinergic nuclei (including the pedunculopontine nucleus and dorsal motor nucleus of the vagus) (
15) are usually demonstrable postmortem in PD. It has been suggested that a cholinergic deficit in visual association areas may underlie VHs. The posterior region of the nbM (predicted to project to the posterior and temporal cortices) is significantly depleted of neurons in PD patients without cognitive deficit when compared with age-matched control subjects (
16). These findings are in keeping with MRI tractography demonstrating a higher degree of abnormality of nbM fiber tracts projecting to posterior cortices in PD patients with VHs (
17).
We analyzed postmortem brain tissue to examine the relationships between VHs, markers of perfusion and cholinergic input to visual processing areas V2 (BA 18) and V3 (BA 19), in PD with and without VHs. We hypothesized that VHs would be associated with biochemical evidence of chronic hypoperfusion (relative to metabolic demand) in visual processing areas V2 and V3 and/or reduced cholinergic input to these areas. To assess this we measured the ratio of MAG to PLP1, as MAG is highly sensitive to reduced tissue oxygenation in chronic hypoperfusion, whereas PLP1 is relatively preserved (
18). In addition, we measured von Willebrand factor, a marker of microvessel density, and vascular endothelial growth factor A (VEGF-A), the production of which is stimulated by hypoxia-inducible factor when tissue oxygen levels fall. We also hypothesized that cholinergic input would be reduced in those with PD and VHs. To assess this, we assayed levels of choline acetyltransferase (ChAT), acetylcholinesterase (AChE), and butyrylcholinesterase (BChE). ChAT is the enzyme that synthesizes acetylcholine (Ach); AChE and BChE are responsible for its breakdown, although BChE primarily metabolizes ACh at higher concentrations.
Methods
Study Cohort
The postmortem cohort for this study was chosen from the Parkinson’s U.K. Tissue Bank and was previously examined by an author (A.L.) (
19). All PD study subjects had neuropathologically confirmed Lewy body pathology. Medical records were reviewed to stratify the cohort into PD, PD with mild cognitive impairment, and PDD groups and to obtain clinical data (e.g., presence or absence of hallucinations). PD study subjects with cognitive deficits severe enough to interfere with independent activities of daily living, satisfying DSM-IV and ICD-10 clinical criteria for dementia and Movement Disorder Society Task Force diagnostic criteria for PDD (
1), were classified as PDD (
1,
20,
21). To distinguish PDD from DLB, the 1-year rule was applied (
22).
Those with significant coexisting neuropathology, including a Braak tangle stage score ≥3, cerebral macro- or microinfarcts, or foci of hemorrhage, were excluded. The control group previously used by Liu et al. (
19) was supplemented with additional control subjects from the South West Dementia Brain Bank due to limited control subject availability from the Parkinson’s U.K. Tissue Bank. The control subjects had no history of cognitive problems and had no neuropathological abnormalities on postmortem examination other than mild neurofibrillary tangle pathology (Braak tangle stage ≤3) or scattered diffuse Aβ plaques in some cases. The PD groups were matched for age, gender, and postmortem interval. Each brain in the PD groups was matched to two control subjects. Ethical approval was granted by the South West Dementia Brain Bank and Parkinson’s U.K. Tissue Bank. Details of sample preparation are presented in the
online supplement.
ELISAs and von Willebrand Factor Dot Blot
As previously described in detail (
18), MAG was assayed by indirect enzyme-linked immunosorbent assay (ELISA), developed in our laboratory. VEGF-A was assayed using a commercial kit (Duoset, DY293B, R&D Systems, Minneapolis) adapted for use in a 384-well plate. von Willebrand factor was assayed by dot blot developed in our laboratory (for further details, see the
online supplement). PLP1 was assayed using a commercial sandwich ELISA kit (Cloud Clone Corp., Wuhan), as per the manufacturer’s instructions. Samples were diluted 1:10 for this assay. Three samples were carried over on each plate to check that the results were consistent. The α-synuclein was measured in soluble and insoluble extracts by ELISA, as described previously, adapted for a 384-well Nunc MaxiSorp plate (
18). ChAT activity and concentration were assayed using an assay developed by Dr. Darreh-Shori’s group at the Karolinska Institute, as described in detail previously (
18,
23).
Field Fraction Analysis for α-Synuclein
Full details of the immunolabeling method are presented in the
online supplement. The number of Lewy bodies per field was counted in 15 fields of BA 18 and BA 19 under a ×20 objective. Image Pro Plus (Media Cybernetics, Rockville, Md.) was used to select the fields at random in the predefined area and for image capture. Each field per section had equal weighting in subsequent analyses.
Statistical Analyses
All ELISAs were adjusted for total protein content. Data were analyzed using parametric statistical tests wherever possible. If a variable was not normally distributed and it was not possible to achieve a normal distribution by transformation, nonparametric tests, such as the Kruskal-Wallis test, were used. Because the postmortem interval was significantly longer in the control group, postmortem interval was included as a covariate in the parametric tests. When Dunn’s test was used (for post hoc comparisons between groups), Bonferroni correction was applied. Correlations for nonparametric variables were assessed using Spearman’s rank correlation coefficients.
The primary outcome measure was a difference in the MAG:PLP1 ratio between groups. Because our hypothesis concerned the presence or absence of VHs in PD (irrespective of the presence or absence of cognitive impairment), the two groups containing individuals with PD who had experienced VHs were combined for analysis. The cohort was further divided in a sensitivity analysis to examine separately those with PD and VHs with and without dementia. A threshold p value of 0.05 was used throughout. By using information from our previous study examining control subjects compared with those with DLB, we estimated that the present study had 84% power to detect a difference of 0.7 in the MAG:PLP1 ratio (
18).
Results
We were able to obtain tissue from 32 control subjects, 11 individuals with PD who did not experience VHs (PD without VHs), and 26 individuals with PD who did experience VHs (PD plus VHs). Sixteen of the individuals with VHs had been diagnosed as having PDD. Information on clinical features was obtained from the patients’ antemortem clinical records. The control subjects had a longer postmortem interval (
Table 1), a difference driven mainly by the short postmortem interval of many of the PD cases, which we could not match in the available control subjects. Postmortem interval was therefore included as a covariate in subsequent analyses. The majority of individuals with VHs had frequent hallucinations. There was no marked difference in disease duration between the PD plus VHs group and PD without VHs group. Unfortunately, information on use of cholinesterase inhibitors was not available.
Table 1 summarizes the characteristics of the PD and control groups, including demographic data, age at death, cause of death, neuropathological diagnosis, Braak Lewy body stage, postmortem interval, and (as applicable) disease duration and frequency of VHs (rated on a 3-point scale: 0=none, 1=infrequent, 2=frequent). Results of a sensitivity analysis subdividing the PD cohort with VHs into those with and without dementia are summarized in
Table 2.
Biochemical Evidence of Hypoperfusion
There was no consistent evidence from the biochemical analyses of chronic hypoperfusion in those with VHs (for further details, see also Figure S1 and Table S1 in the
online supplement). Indeed, in several brain areas there was evidence that perfusion might have been increased in the VHs group. Neither was there any evidence of a difference between the two PD groups in the markers of perfusion. There were no between-group differences in VEGF-A in any of the brain areas under investigation. Microvessel density was decreased in the PD plus VHs group in both ventral BA 18 (p=0.004) and BA 19 (p=0.001) but not in either dorsal brain area (see Figure S1 and Table S1 in the
online supplement). There was weak evidence that von Willebrand factor tended to fall as α-synuclein concentration increased in ventral BA 18 and BA 19 (see Figure S4 in the
online supplement).
α-Synuclein.
As expected, insoluble α-synuclein was increased in both PD groups (see Table S2 and Figure S2 in the
online supplement). There was no evidence of a between-group difference in Lewy body counts. Visual inspection of the slides suggested increased finely granular cortical immunopositivity for α-synuclein in the PD groups, rather than a larger number of discrete Lewy bodies. Similarly, Braak Lewy stages were very similar for the PD without VHs and PD plus VHs groups (
Table 1).
Biochemical Evidence of Disturbance of the Cholinergic Equilibrium
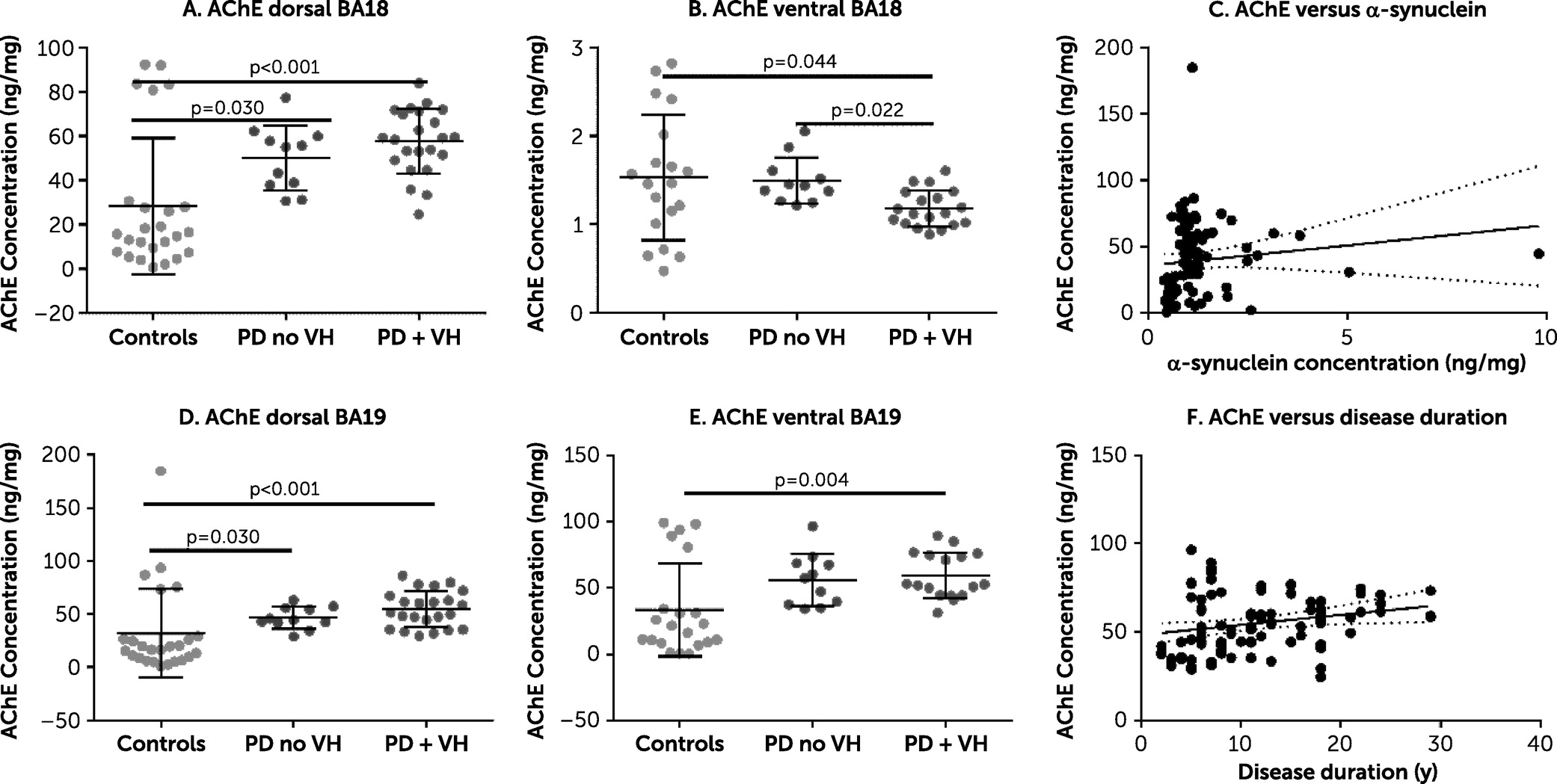
There was strong evidence of a between-group difference in AChE. Specifically, AChE was increased in the PD plus VHs group in three of the four brain areas under investigation (Cohen’s d=0.71–1.22) (
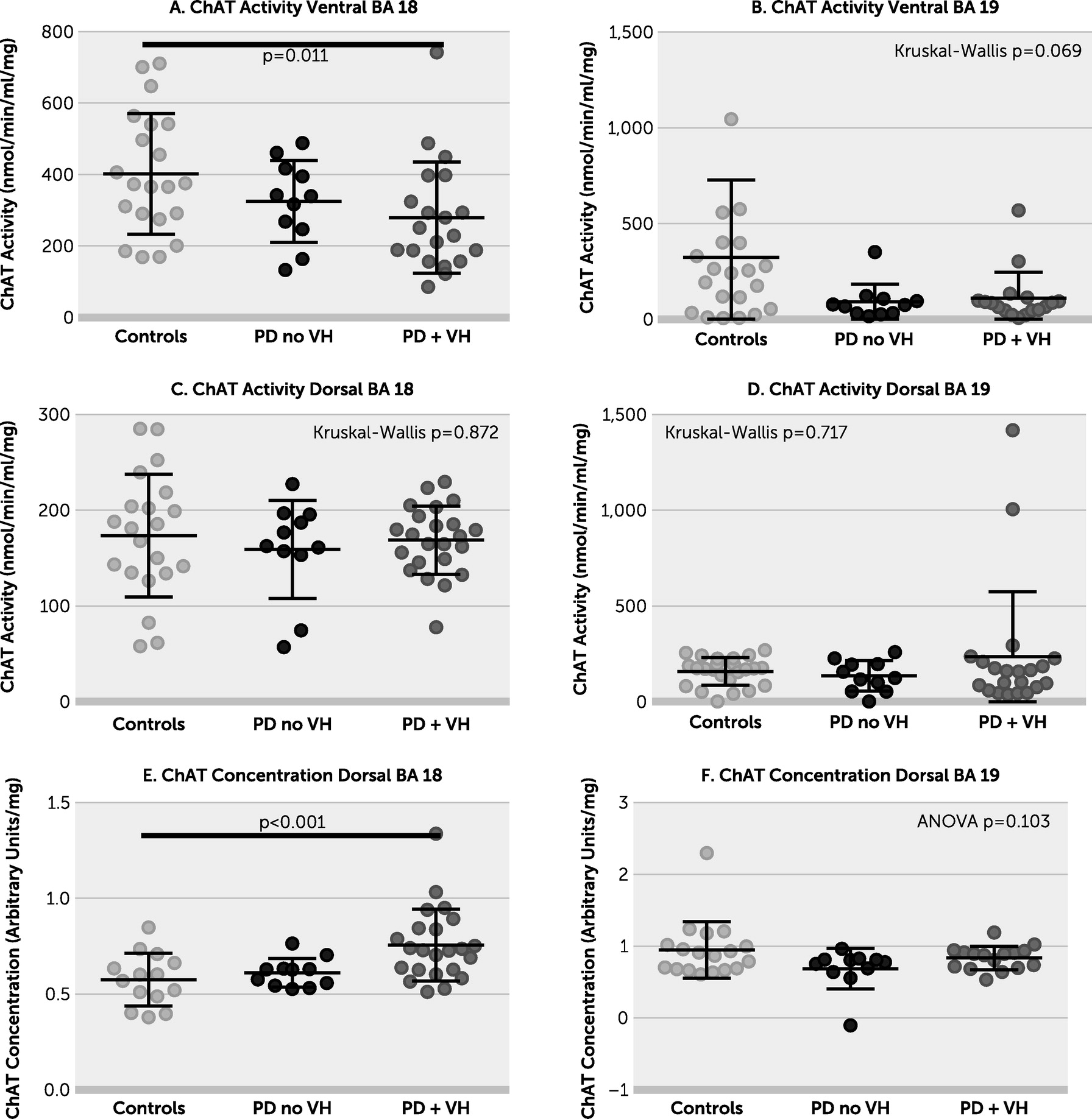
Figure 1). The lack of difference between the PD plus VHs group and PD without VHs group in most brain areas under investigation may be due to low numbers or a true lack of effect. There was no evidence of a relationship between AChE and either duration of PD or concentration of insoluble α-synuclein. ChAT concentration was increased in the PD plus VHs group in dorsal BA 18 (
Figure 2; see also Table S3 in the
online supplement) but not in dorsal BA 19. ChAT activity was decreased in the PD plus VHs group in ventral BA 18, the pattern of data resembling that of AChE concentration in the same region. There was weak evidence of a between-group difference in ChAT activity in ventral BA 19; ChAT activity declined in ventral BA 19 with increasing AChE (see Figure S3 in the
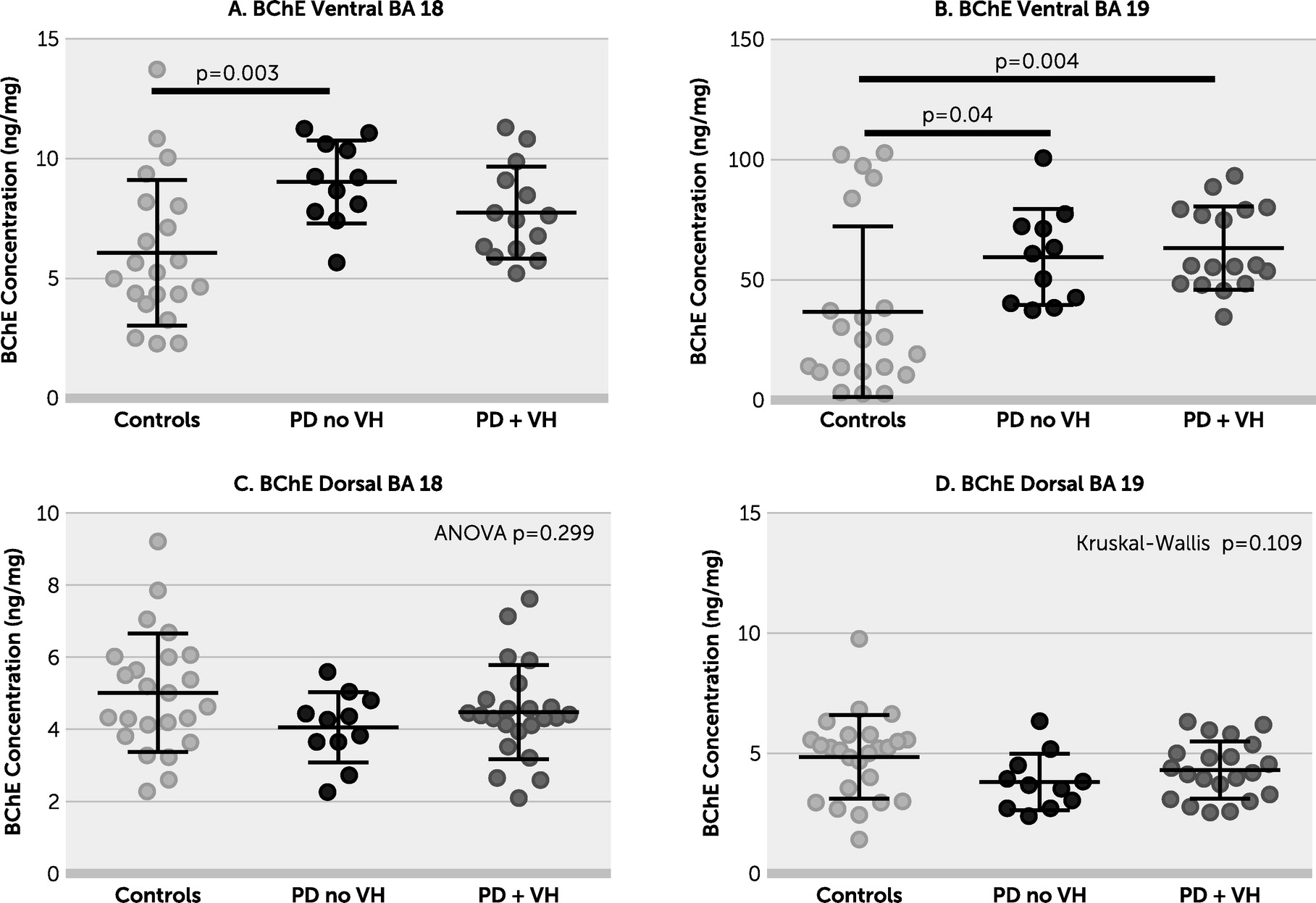
online supplement). There was no evidence of a between-group difference in ChAT activity in dorsal BA 18 or BA 19. BChE was increased in the PD without VHs group in ventral BA 18 and in both PD groups in ventral BA 19. There were no between-group differences in BChE in either dorsal brain area (
Figure 3).
As shown in Figure S4 in the
online supplement, AChE in dorsal BA 18 and BA 19 was closely correlated, but this was not the case in the ventral brain areas. There was a positive correlation between AChE and the MAG:PLP1 ratio in dorsal BA 19 (Spearman’s rho=0.65, p<0.001) but not in dorsal BA 18, where there was no clear relationship (Spearman’s rho=0.012, p=0.93). AChE was not related to insoluble α-synuclein concentration.
Discussion
Despite adequate power, we found no strong evidence of chronic hypoperfusion in individuals with PD with or without VHs in any of the brain areas examined. This is consistent with our previous study in individuals with Alzheimer’s disease with or without VHs and individuals with DLB (
18). In the sensitivity analysis there was little difference in the findings when the groups were subdivided according to the presence or absence of dementia (see Table S4 in the
online supplement). As expected, insoluble α-synuclein was increased in both PD groups in all four brain areas under investigation, but levels did not differ between the PD plus VHs group and PD without VHs group. The decrease in microvessel density in ventral BA 18 and BA 19 is consistent with our previous findings in DLB (
24). The increase in AChE may have improved perfusion by causing vasodilatation, perhaps explaining the decreased von Willebrand factor seen in both ventral brain areas in the PD plus VHs group (see Figure S1 in the
online supplement).
There was evidence of a change in the cholinergic equilibrium in BA 18 and BA 19 in PD, most marked in the PD plus VHs group. AChE metabolizes ACh at lower concentrations than BChE, which works more efficiently at higher concentrations. We found no evidence of a between-group difference in BChE, but did find that AChE was increased in the PD plus VHs group in three out of the four brain areas under investigation. This suggests that any differences in cholinergic activity occur at lower concentrations of ACh. A small number of case subjects in the PD plus VHs group took AChE inhibitors, which may have led to an increase in AChE. BChE expression has been demonstrated in multiple human brain regions, including in areas where it appears to be coexpressed with AChE. The extent of expression of both enzymes in normal human occipital cortex is unclear.
Our findings on assessment of markers of occipital perfusion differ from those of some in-vivo imaging studies. Although this may reflect postmortem changes, or that the MAG:PLP1 ratio is a marker of perfusion over a relatively long period of time, it is noteworthy that the imaging studies have themselves yielded quite varied findings. In-vivo imaging studies have also examined cholinergic innervation in PD. An early SPECT study that mapped the vesicular ACh transporter found reductions in this transporter in multiple cortical regions in PDD compared with PD without dementia (
25). Two relatively small PET studies showed reduced AChE activity in those with PDD, particularly in the occipital lobe (
26,
27). The changes were much less pronounced in PD without dementia. Klein et al. (
27) commented that cholinergic pathology seemed more pronounced in posterior regions of the brain. A larger PET study which measured AChE activity using [
11C]MP4A found a reduction in AChE activity early in PD, particularly in BA 18. The authors also found more widespread, profound reductions in AChE activity in PDD compared with advanced PD without dementia (
26).
Our finding of alterations in ChAT in both dorsal and ventral BA 18, although in opposing directions, provide further evidence of changes in the cholinergic equilibrium in these study subjects. Several previous studies have demonstrated a loss of ChAT activity in PD and DLB. Tiraboschi et al. (
28) found a reduction in ChAT activity in midfrontal and hippocampal tissues, with the reduction being more marked than that in Alzheimer’s disease. A much smaller study found reductions in ChAT activity in several thalamic nuclei in PDD, but less so in PD without dementia (
29). A study of the relationship between Lewy bodies and ChAT activity found large reductions in ChAT activity in the hippocampus and the prefrontal and temporal cortex that tended to correlate with the number of Lewy bodies in these areas (
30). Similarly, Harding et al. (
31) found that VHs correlated with Lewy body burden in the temporal lobe. Mattila et al. (
30) demonstrated that the decrease in frontal ChAT activity was greatest in those with PD and cognitive impairment. A small study in the 1980s found that ChAT activity was reduced in multiple cortical areas in PD (compared with control subjects), although the evidence for a reduction in the occipital cortex was weak (
32). A previous, smaller, study of individuals with DLB and Alzheimer’s disease found ChAT activity to be reduced in BA17 and BA 18/19 in DLB (
33).
Several previous studies have demonstrated that cholinergic innervation of neurons in the occipital cortex influences visual attention and responses. It seems therefore likely that deficient cholinergic input would influence visual processing, although it was previously suggested that visual areas V1-V3 are less likely to be implicated in VHs in PD, possibly due to preservation of these structures (
34).
Strengths of this study include the well-characterized cohort, relatively short postmortem intervals, separate analysis of ventral and dorsal visual processing areas, assessment of cholinergic catabolic enzymes, as well as ChAT, and the large number of control subjects. Weaknesses include the relatively small number of PD without VH cases, lack of measurement of other neurotransmitters (e.g., dopamine), limited information on the nature of the VHs, inability for technical reasons to assay ChAT concentration in ventral BA 18 and BA 19, and slightly higher postmortem interval in the control subjects.
In summary, we report that there are changes in the cholinergic equilibrium in BA 18 and BA 19 in PD that are particularly marked in PD plus VHs. This raises the possibility that treatments for VHs in PD should be targeted to the cholinergic system, but further research is required to verify these findings. Future research should also assess other components of the visual processing pathway (e.g., V4–V6).