Case Presentation
A 60-year-old right-handed man, who was a retired electrical engineer, presented to our clinic after 2 years of insidious-onset progressive word-finding difficulties, as well as an observed decline in verbal output (
1). He described his symptoms as “trouble capturing thoughts in my mind” and “losing words in my head.” He reported problems with complex decision making and denied difficulties with language comprehension, memory, and tasks involving visuospatial reasoning. He and his wife noted mild social withdrawal, increased frustration, and “trying less” in most activities over the previous 2 years. There were no personality changes, signs of current major depression, or significant apathy. He was successfully driving, playing golf, and managing household finances and otherwise independent in basic and instrumental activities of daily living (ADLs).
The patient’s past medical history included hypertension and major depressive disorder. His medications included olmesartan and escitalopram. Social and developmental history were negative for dyslexia and other learning disabilities. He completed college with a degree in electrical engineering and worked as a systems consultant, retiring 3 years prior to symptom onset at age 55, with no cognitive difficulties at work. There was no history of substance use. His family history was notable for Parkinson’s disease in his brother and coronary artery disease.
Question: What are diagnostic considerations based on history alone?
The history of insidious onset of cognitive (language) symptoms, which worsened progressively over 2 years in our patient, is most suggestive of a neurodegenerative disease. Alzheimer’s disease and the frontotemporal dementias are the most common diseases when symptom onset occurs before age 65. Early-onset Alzheimer’s disease is more likely than late-onset Alzheimer’s disease to present with primary cognitive dysfunction in nonmemory domains, such as language or executive function (
2). Neuroimaging will rule out unlikely causes like an unusual, slow-growing focal lesion that could cause highly circumscribed verbal deficits.
Neurocognitive assessment of affected domains of cognition and behavior allows us to characterize symptoms according to known cognitive syndromes, initially in a “neuropathology blind” way. Identifying a known cognitive syndrome allows the initial generation of a neuropathologic differential diagnosis based on already identified clinical-pathologic correlates.
Assessing the degree to which symptoms affect daily functioning can help differentiate between prodromal and later stages of neurodegenerative disease. Our patient presented with intact functioning in his instrumental and basic ADLs and with objective impairments on cognitive testing, placing him at the level of mild cognitive impairment, synonymous with mild neurocognitive disorder. If he had no objective deficits, his syndrome would have been considered subjective cognitive decline, which in some instances is a risk factor for developing mild cognitive impairment and later impairments in daily functioning (
3).
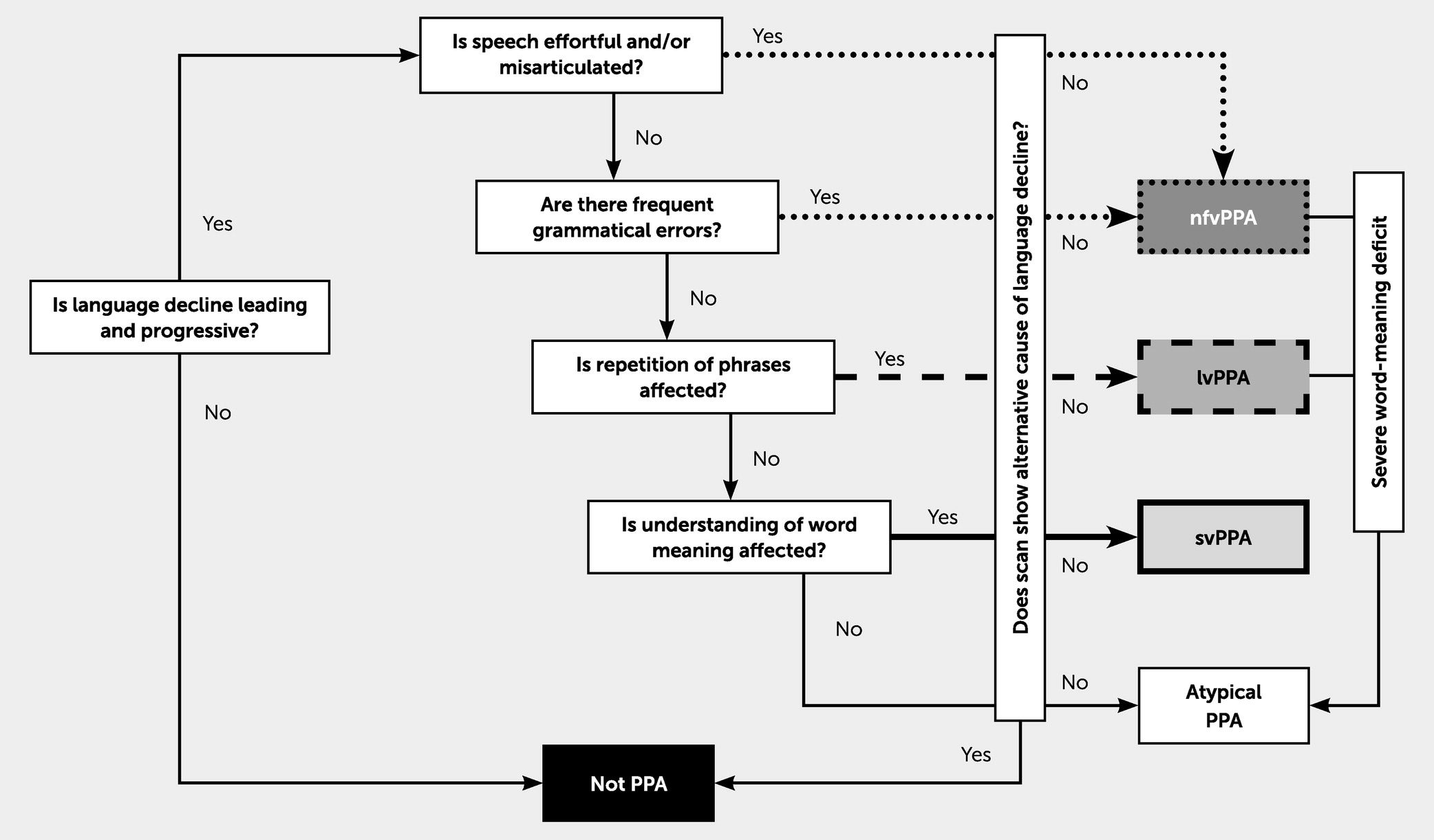
Our patient’s primary reported complaints were in the domain of language, with a history that suggests relative preservation of memory, attention, executive function, and visuospatial abilities. Generally, history of initial decline in one or more domains of language production is concerning for primary progressive aphasia (PPA), a group of degenerative language syndromes for which clinical-radiologic-pathologic comparative studies have identified three variants with formalized diagnostic criteria and difficult-to-classify cases variously called “atypical” or “mixed” (
4–
6). Variants differ in aspects of language affected and in most common underlying neuropathology.
The three canonical variants include nonfluent-agrammatic variant PPA (nfvPPA), semantic variant PPA (svPPA), and logopenic variant PPA (lvPPA) (
4–
6). In nfvPPA, defining linguistic deficits include agrammatism, effortful and halting speech (with phrase length typically less than six words) with or without apraxia of speech, and neuroanatomic correlates of atrophy and/or hypometabolism in the left inferior frontal lobe, often in the posterior fronto-insular cortex. In svPPA, defining linguistic deficits include impaired single-word comprehension and naming, with typical neuroanatomic correlates of bilateral and left-greater-than-right atrophy and/or hypometabolism in the anterior temporal lobes. In lvPPA, defining linguistic deficits include impaired word retrieval and phrase or sentence repetition, with left posterior perisylvian/parietal atrophy and/or hypometabolism (
6–
9). There is heterogeneity within subtypes and ongoing debate regarding core defining criteria (
9,
10).
Figure 1 depicts a simplified, diagnostic decision tree (
8).
Up to 40% of cases may not clearly meet consensus criteria for the three canonical variants, and symptoms may be “mixed” across variants or are “atypical,” involving specific language dysfunction not represented in criteria for canonical variants (
11–
13).
Question: How does the neurocognitive and physical examination contribute to our understanding of diagnostic considerations?
Neurocognitive testing within the language domain begins with observation of spontaneous language output to assess fluency, grammar, word-finding pauses, and speech errors. Formal assessment of speech praxis, expressive language output, verbal fluency (letter- and category-based), motor speech, naming, repetition, and comprehension follows. At initial evaluation, 2 years into the disease course, our patient’s language impairment was notable for variability in word-finding pauses and overall sparse speech output, especially with spontaneous narrative generation. His speech was grammatical, with no apraxia of speech or dysarthria. Verbal fluency was impaired, with phonemic fluency weaker than category-based fluency (for further details, see video S1 in the
online supplement). Design fluency was relatively intact, suggesting that verbal fluency deficits are more related to language impairment than to executive dysfunction. Confrontation naming was also relatively intact, despite word-finding pauses. Repetition, partially dependent on auditory verbal working memory, and basic comprehension were intact. The patient displayed intact decoding, reading comprehension, and written language abilities. Additionally, there were limitations in his attention and executive functioning (see Tables S1 and S2 in the
online supplement).
Elemental neurologic examination had notable absence of involuntary movements, dystonia, Parkinsonism, and changes in extraocular motility. If present, these findings increase suspicion for underlying frontotemporal lobar degeneration (FTLD), especially progressive supranuclear palsy (PSP) and corticobasal (CBD) degeneration.
The patient’s examination thus far revealed a salient language output deficit and a normal neurologic examination. Given worsening of language impairment over time, with relative preservation of other cognitive domains, his syndrome was most suggestive of a PPA. Considering the canonical PPA variants, his pattern of fluent grammatical speech with intact motor aspects of speech was not suggestive of nfvPPA. Intact confrontation naming and sentence repetition made lvPPA less appropriate, even when applying an expanded conception to allow for normal repetition and prominent word retrieval and naming deficits (
14). Furthermore, his intact naming and single-word comprehension (as shown through his ability to name pictures, a task that indexes semantic memory) were incompatible with svPPA. Symptoms did not meet consensus criteria, nor did they meet expanded conceptions for the three canonical PPA variants, “mixed” PPA, or “anomic” PPA (
4,
6,
15). We could conceptualize this case as an “atypical” or “unclassifiable” PPA (
8,
16,
17). However, this specific syndrome has been previously described in the context of neurodegeneration and of lesions to the left prefrontal cortex from neoplasm and infarction and termed “dynamic” or transcortical motor aphasia (
1,
18).
Question: Can we more specifically localize the deficits? What additional tests or studies are indicated? How do these tests guide prediction of clinical-pathologic correlation?
Considering the examination findings of a relatively circumscribed language output deficit, we hypothesized a disruption in the region of the superior frontal gyrus (SFG) and its connections, which are involved in recruitment of attention and executive control networks necessary for initiation of language (
19). The SFG includes the supplementary motor area, which—when on the language dominant hemisphere—is thought to subserve initiation/activation, planning, and coordination of language via a fiber tract to the inferior frontal gyrus (
20). Given findings of preserved grammar, motor aspects of speech, and auditory verbal working memory, we might expect sparing of the inferior frontal gyrus, insula, temporal lobes, and white matter tracts connecting these regions (
7,
8).
Neuroimaging can be helpful to interrogate hypothesis-driven anatomical localization that explains observed patterns of impairment. Brain MRI allows for visualization of focal or segmental patterns of atrophy. The clinical read of our patient’s initial brain MRI suggested minimal global volume loss, with slight predominance in the left frontal and parietal cortices (see Figure S1 in the
online supplement). Our initial follow-up FreeSurfer-calculated cortical thickness analysis demonstrated reduced volume in left-greater-than-right midcaudal superior frontal and caudal middle frontal gyri, compared with 20 healthy, age-matched control subjects (
1). Additional analyses concluded that volume loss was isolated to the left SFG.
Positron emission tomography (PET) imaging using intravenous injection of [
18F]fluorodeoxyglucose (FDG-PET) may provide further information about functional abnormalities, which are often observable prior to the development of brain atrophy (
21). Distinct hypometabolic patterns have been observed for the three canonical PPA variants and may also advance neuropathologic differential diagnosis (
6). Brain single-photon emission computed tomography (SPECT) also could be considered. While some studies demonstrate higher sensitivities and specificities for FDG-PET than SPECT, others have shown equivalence, and SPECT may be a more accessible test to obtain in some centers (
22,
23).
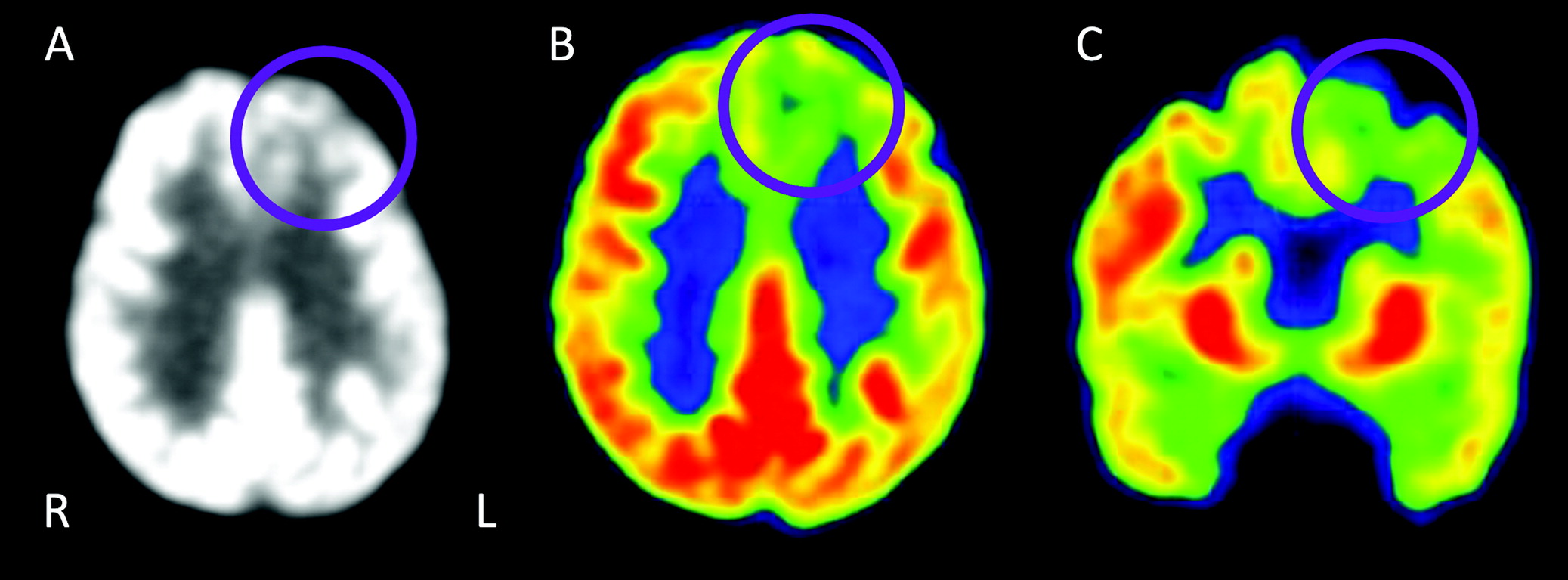
Our patient’s FDG-PET scan demonstrates hypometabolism in the left SFG, with preserved metabolism elsewhere (
Figure 2), consistent with the early atrophy pattern seen on MRI and with the patient’s syndrome. While this is not a typical pattern for a canonical PPA syndrome, isolated frontal lobe hypometabolism is most suggestive of FTLD pathology.
Other currently available tests that could be considered are amyloid- and tau-based molecular PET scans and cerebrospinal fluid (CSF) biomarkers, which are used primarily in clinical settings to detect Alzheimer’s disease pathology. We obtained Alzheimer’s disease CSF biomarkers to guide more predictive clinical-pathologic correlation given the atypical PPA variant, early age at onset, and absence of other examinations or neuroimaging results.
CSF analysis by enzyme-linked immunoassay for beta-amyloid 1-42 (Aβ42), total tau (t-tau), and phosphorylated tau181 (p-tau181), which are associated with Alzheimer’s disease pathology, revealed Aβ42 of 980 pg/mL, t-tau of 201.2 pg/mL, and p-tau181 of 45.8 pg/mL. The levels of each protein were within normal limits (
24). We still lack robust CSF biomarkers to identify forms of FTLD, the most common subtypes of which involve pathological inclusions of the protein tau (i.e., FTLD-tau) or the protein transactive response DNA-binding protein 43 kDa (TDP-43) (i.e., FTLD-TDP). Primary tauopathies may or may not show elevations in total CSF tau.
Comparing findings to those of the described PPA syndromes and their common underlying pathologies can help narrow the neuropathological differential diagnosis in an atypical case. Any PPA case can be caused by single or multiple pathologic subtypes (
9,
25). The most common neuropathologies that underly PPA are FTLD (associated with nfvPPA and svPPA) and Alzheimer’s disease (associated with lvPPA). Dementia with Lewy bodies (DLB) and prion disease are rare causes (
26,
27). In our patient, Alzheimer’s disease pathology was unlikely given the distribution of atrophy and hypometabolism, as well as the results of CSF Alzheimer’s disease biomarkers. There were no clinical or radiologic features suggestive of DLB or prion disease; FTLD was the most likely pathology.
Cases of dynamic aphasia have been associated with the classical clinical syndrome of PSP (Richardson syndrome), as well as with neuropathologic findings of FTLD-PSP, a pathologic subtype of FTLD-tau (
28–
30). However, as noted above, our patient lacked secondary motor symptoms and signs suggestive of FTLD-PSP. A strongly asymmetric pattern of frontal hypometabolism with FDG-PET has been associated with FTLD-tau subtype Pick’s disease (FTLD-PiD), though many cases involve both frontal and temporal cortices (
31,
32).
Question: Considering these additional data, what would be an appropriate diagnostic formulation?
Here, we have a case of insidious, slowly progressive language impairment, with salient deficits in verbal output, consistent with prior descriptions of “dynamic” aphasia, an “atypical” PPA syndrome, at the severity level of mild cognitive impairment.
Symptoms and examination findings broadly suggest disruption of the left dorsal prefrontal cortex, and imaging and biomarkers more precisely support this localization, revealing focal MRI atrophy and circumscribed FDG-PET hypometabolism in the left SFG. Given overlap in the anatomic distribution of “dynamic aphasia” and nfvPPA (which is most often caused by FTLD-tau), we predict underlying FTLD-tau neuropathology, either FTLD-PSP or FTLD-PiD subtypes.
Question: How does longitudinal course of the illness inform predictive clinical-pathologic correlation of underlying neuropathological processes?
The disease ran its course over 12 years. Three years after symptom onset, the patient’s speech became more sparse and executive functioning and attention deficits more profound. This suggests pathologic spread—likely anteriorly and inferiorly—to areas further subserving speech output and language and executive control networks (
19,
20,
33,
34). Concurrently, the patient started to experience frustration and anger more easily and was noted to be more socially withdrawn. Serial MRI reflected progression of atrophy to involve bilateral frontal, temporal, and parietal lobes, with some predominance in left-greater-than-right frontal and temporal lobes (
Figure 3).
By year 6, his symptoms progressed to include apraxia of speech and other features seen in nfvPPA. His speech was more effortful, more dysfluent with sound errors, and developed a “groping” quality. He started to have grammatical errors, first in writing and then in speech. Comprehension of syntactically complex material, naming, and repetition also declined. Behaviorally, he developed symptoms that overlap with behavioral variant FTD (bvFTD) syndrome, including apathy, hyperorality, and disinhibition (
31). He became dependent in instrumental ADLs.
MRI in year 8 demonstrated profound frontal lobar atrophy, mildly asymmetric (left greater than right), with “knife-edge” configuration of gyri (
Figure 3C). In year 9, the patient could not repeat his name and was dependent in basic ADLs. Two years later, he developed extrapyramidal symptoms and was moved to a nursing home. By year 12, he was mute and exhibited no ability for language comprehension. During that year, he contracted COVID-19 and died of complications related to this illness.
Considering the evolving features of nfvPPA and bvFTD our patient developed, it is common in FTDs to see syndromic overlap, especially with disease progression. Common neuropathological associations with nfvPPA include different subtypes of FTLD-tau (CBD, PSP, and PiD) and FTLD-TDP type A. That he did not develop other neurologic signs or symptoms commonly seen in PSP or CBD reduces the likelihood of underlying FTLD-PSP or FTLD-CBD. While not exclusive to one pathology, the aforementioned pattern of atrophy with asymmetrical bilateral frontal, temporal, and parietal atrophy and gross “knife-edge” appearance is most consistent with the evolution of atrophy seen in FTLD-PiD (
32,
35). Taken all together, these syndromic and imaging changes, over time, are most suggestive of the underlying FTLD-tau subtype FTLD-PiD.
Discussion
Dynamic aphasia has been described as an impairment along the “thought-verbal interface,” where there is difficulty in translating one’s thoughts into verbal output (
18,
33,
39). In our patient, difficulty with word retrieval and intact naming performance suggests dysfunction in executive components of language, such as organizing thoughts into expression. This case validates previously identified structure-function relationships linked to dynamic aphasia and provides insight into initial stages of language generation as being related to broader executive-functioning systems (
19). Specifically, in certain models of executive function and working memory, speech output requires the “motivating” of thoughts into speech, which involves some organization (executive control) of the output of language to accurately represent those thoughts (
1).
Over time, the clinical features in this case evolved to include symptoms also seen in nfvPPA and bvFTD, reflecting spread to widely distributed components of language and frontal executive-mediated functions. Symptom progression correlates with progression of atrophy seen on serial brain MRIs, starting in the left SFG and moving inferiorly, posteriorly, and bilaterally. In contrast to cases that present initially with nfvPPA, the initial locus of neurodegeneration in our patient was in the left SFG rather than the inferior frontal lobe, as is typical in nfvPPA. Over time, it is likely that pathology spread from the superior lobe to the inferior frontal lobe, which accounts for the development of nfvPPA-like symptoms. This pattern of pathologic spread can vary by subtype of PPA and underlying FTLD (
40,
41). As neurodegenerative diseases progress, pathology spreads, and therefore it is useful for clinicians to continue to update their assessments and syndromic classification.
To our knowledge, this is the first report of a primary dynamic aphasia syndrome due to underlying FTLD-PiD. At least two other cases with similar syndromes have been reported to have underlying FTLD-PSP pathology, a 4R tauopathy (
29,
30). PiD is a 3R tauopathy with Pick bodies (characteristic argyrophilic neuronal cytoplasmic inclusions), classified under the pathologic “umbrella” of FTLD-tau. Among the canonical PPA clinical syndromes, FTLD-PiD is less common as the dominant underlying pathology. However, in nfvPPA, the frequency of underlying FTLD-PiD is comparable to that of other pathologies, including CBD, PSP, and FTLD-TDP type A (
9,
42–
44).
There is heterogeneity in the clinical syndromes that have a pathologic diagnosis of PiD. These most commonly include bvFTD, followed by nfvPPA and svPPA, and rarely CBD syndrome and amnestic syndrome (
45–
47). In several series, about 60% of FTD-PiD cases identified as postmortem were diagnosed with bvFTD, while around 40% were diagnosed with PPA (
47,
48). The better we can become at accurate clinicopathologic correlations during life, the better we are positioned to study and, ultimately, treat patients, perhaps even with disease-modifying therapy aimed to ameliorate specific neuropathology.
However, disease biomarkers themselves will not teach us about brain-behavior relationships. Being able to carefully trace the changing distribution of neuropathology and link it to changing clinical symptomatology will add to our understanding of how the brain works. Identifying the initial focality of neurodegenerative diseases and following its spreading distribution over time are worthy activities for neuropsychiatrists and behavioral neurologists.