CONCEPTUAL FRAMEWORK, DEFINITIONS, AND ANIMAL MODELS
Drug addiction, also known as substance dependence, is a chronically relapsing disorder characterized by (1) compulsion to seek and take the drug, (2) loss of control in limiting intake, and (3) emergence of a negative emotional state (e.g., dysphoria, anxiety, irritability) when access to the drug is prevented (defined here as dependence) (
1).
Addiction and
substance dependence (as currently defined by the
Diagnostic and Statistical Manual of Mental Disorders, 4th edition) (
2) will be used interchangeably throughout this paper to refer to a final stage of a usage process that moves from drug use to addiction. Clinically, the occasional but limited use of a drug with the
potential for abuse or dependence is distinct from escalated drug use and the emergence of a chronic drug-dependent state. An important goal of current neurobiological research is to understand the neuropharmacological and neuroadaptive mechanisms within specific neurocircuits that mediate the transition from occasional, controlled drug use and the loss of behavioral control over drug-seeking and drug-taking that defines chronic addiction.
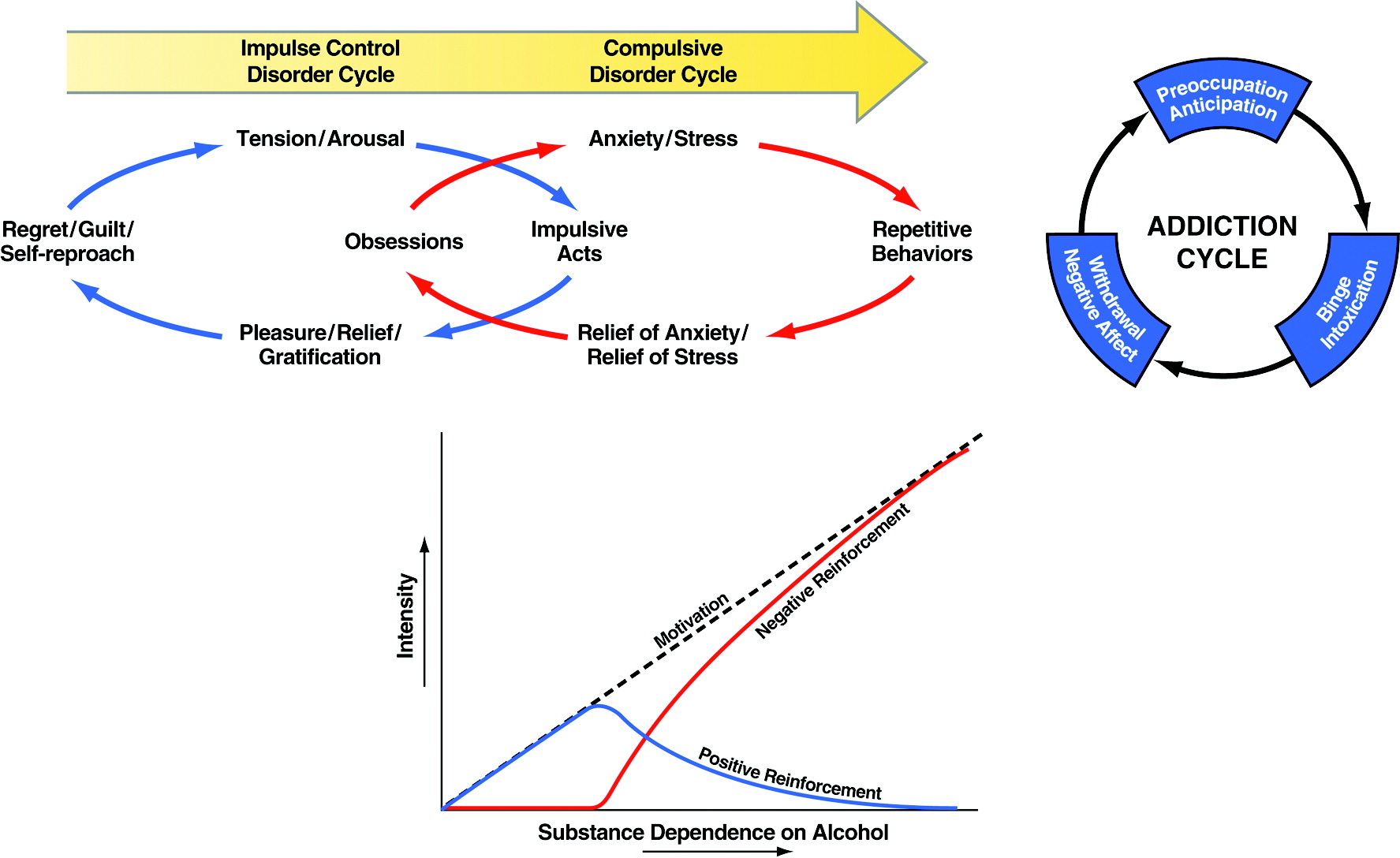
Addiction has been conceptualized as a chronic relapsing disorder with roots both in impulsivity and compulsivity and neurobiological mechanisms that change as an individual moves from one domain to the other (
3). In addiction, drug-taking behavior progresses from impulsivity to compulsivity in a three-stage cycle:
binge/intoxication,
withdrawal/negative affect, and
preoccupation/anticipation. As individuals move from an impulsive to a compulsive disorder, the drive for the drug-taking behavior shifts from positive to negative reinforcement (
Figure 1). Impulsivity and compulsivity can coexist in different stages of the addiction cycle.
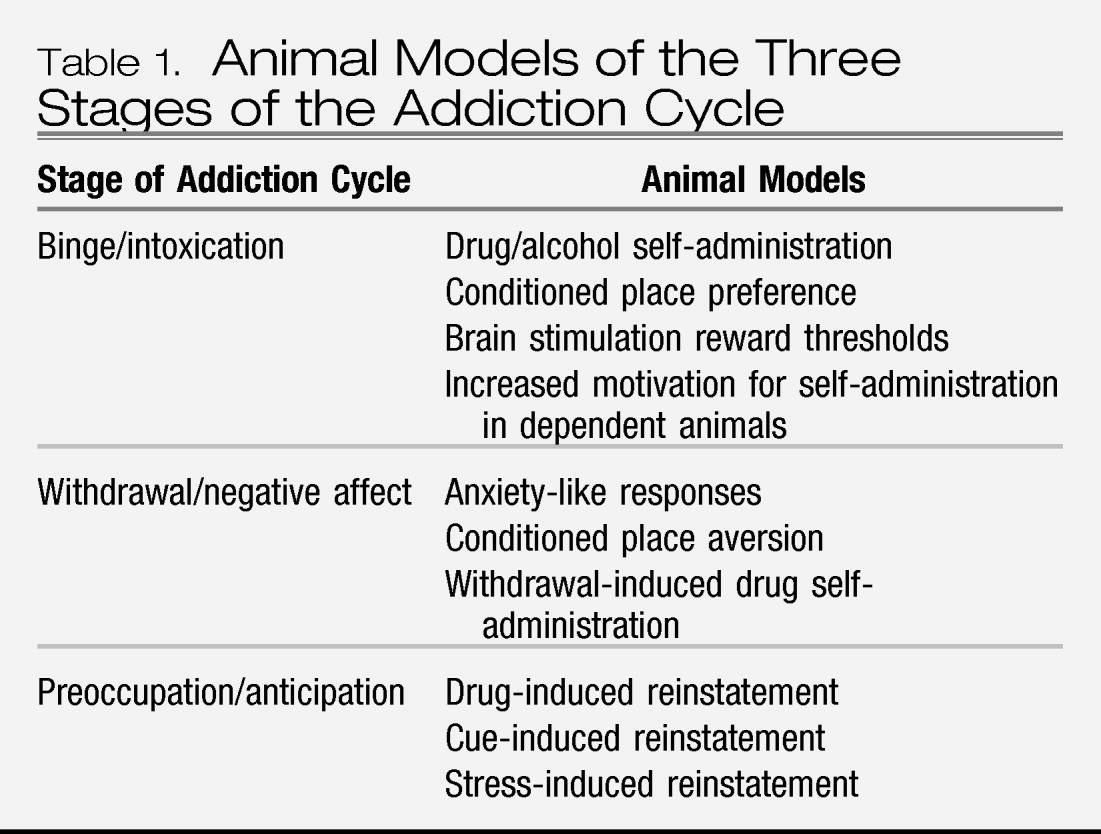
Much of the recent progress in understanding the neurobiology of addiction has derived from the study of animal models of addiction on specific drugs such as opiates, psychostimulants, and alcohol (
4). While no animal model of addiction fully emulates the human condition, animal models do permit investigation of specific elements of the process of drug addiction. Such elements can be categorized by models of different stages of the addiction cycle. While much focus in animal studies has been on the synaptic sites and transductive mechanisms in the nervous system on which drugs with dependence potential act initially to produce their acute positive reinforcing effects (
binge/intoxication stage), new animal models of chronic drug taking and seeking (
withdrawal/negative affect stage) and the craving stage (
preoccupation/anticipation) have been developed and are beginning to be used to explore how the nervous system adapts to drug use (
Table 1). The neurobiological mechanisms of addiction that are involved in various stages of the addiction cycle have a specific focus on certain brain circuits and the molecular/neurochemical changes associated with those circuits during the transition from drug-taking to drug addiction and how those changes persist in the vulnerability to relapse (
5).
NEUROBIOLOGICAL MECHANISMS OF THE BINGE/INTOXICATION STAGE
A long-hypothesized key element of drug addiction is that drugs of abuse activate brain reward systems, and understanding the neurobiological bases for acute drug reward has been a key to how these systems change with the development of addiction (
1,
6). A principle focus of research on the neurobiology of the positive reinforcing effects of drugs with addiction potential has been the origins and terminal areas of the mesocorticolimbic dopamine system, and there is compelling evidence for the importance of this system in drug reward. Much work suggests that activation of the mesocorticolimbic dopamine system has multiple functional attributes, including giving incentive salience to stimuli in the environment (
7) to drive performance of goal-directed behavior (
8) or activation in general (
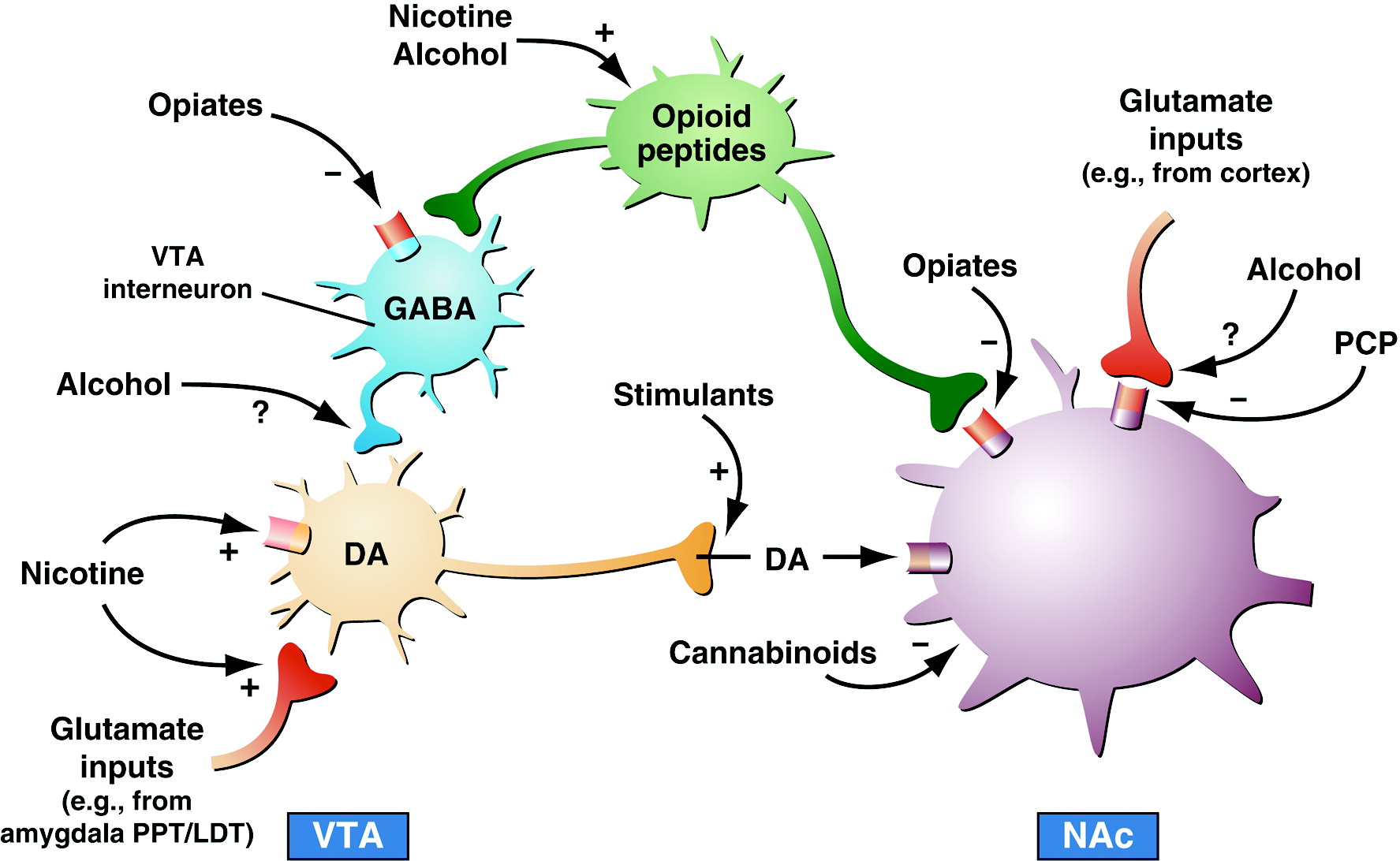
9). However, the specific circuitry associated with drug reward in general has been broadened to include the many neural inputs and outputs that interact with the basal forebrain, specifically the nucleus accumbens (
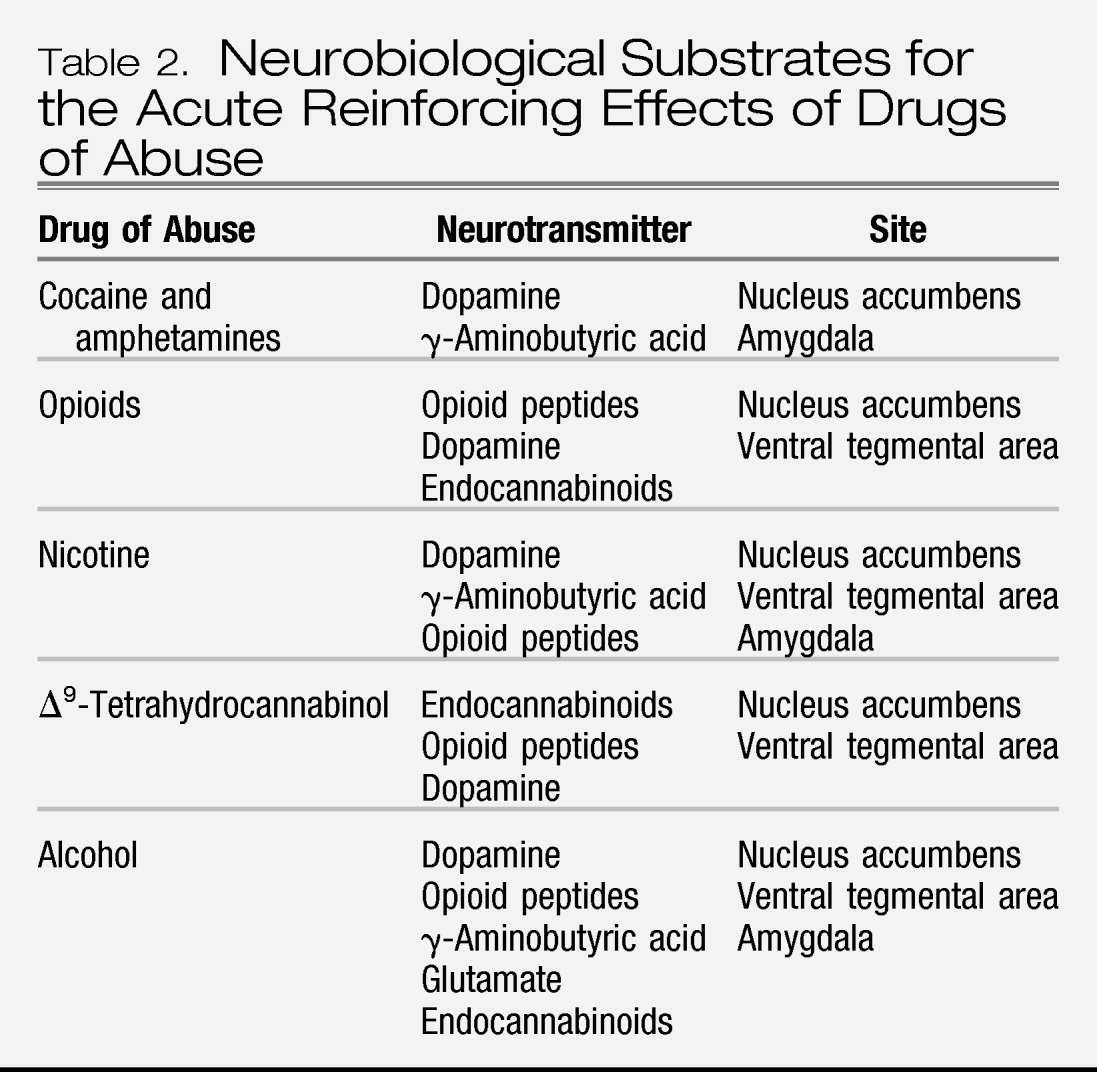
Figure 2). As the neural circuits for the reinforcing effects of drugs with dependence potential have evolved, the role of neurotransmitters/neuromodulators also has evolved, and four of those systems have been identified to have a role in the acute reinforcing effects of drugs: dopamine, opioid peptides, γ-aminobutyric acid (GABA), and endocannabinoids (
Table 2).
The mesolimbic dopamine system is well established as having a critical role in the activating and reinforcing effects of indirect sympathomimetics such as cocaine, methamphetamine, and nicotine. However, while all drugs of abuse acutely activate the mesolimbic dopamine system, particularly in the medial shell region of the nucleus accumbens, the role of dopamine becomes less critical as one moves to opioid drugs, alcohol, and Δ
9-tetrahydrocannabinol (Δ
9-THC). Here, other neurotransmitter systems such as opioid peptides, GABA, and endocannabinoids may play key roles either in series or independent of activation of the mesolimbic dopamine system. Other components of the basal forebrain that have been identified with drug reward have also focused on the amygdala (
5,
10). For example, a particularly sensitive site for blockade of the acute reinforcing effects of alcohol with opioid and GABA-ergic antagonists appears to be the central nucleus of the amygdala (
11). Opioid peptide antagonists also block the reinforcing effects of Δ
9-THC, a key active ingredient in marijuana. Cannabinoid CB
1 antagonists block opioid, alcohol, and cannabinoid reward (
12,
13). In summary, all drugs of abuse activate the mesolimbic dopamine system, but much evidence suggests that dopamine-independent reinforcement occurs at the level of the nucleus accumbens, suggesting multiple inputs to the activation of critical reinforcement circuitry in these brain regions (
14,
15). Thus, multiple neurotransmitters are implicated in the acute reinforcing effects of drugs of abuse. Key players in the nucleus accumbens and amygdala are dopamine, opioid peptide, and GABA systems with modulation via endocannabinoids.
Other elements of the acute drug reward circuit include the ventral pallidum and dorsal striatum. A major output from the nucleus accumbens is to the ventral pallidum/substantia innominata, and elements of the ventral pallidum may not only be critical for further processing of the drug reward signal but also may be directly modulated by drugs of abuse (
16,
17). The dorsal striatum does not appear to play a major role in the acute reinforcing effects of drugs of abuse but appears to be recruited during the development of compulsive drug seeking (
18), suggesting that the dorsal striatum may play a minor role in the acute reinforcing effects of psychostimulant drugs but a key role in the transition to compulsive use (
18).
In summary, much is known about the neurobiological circuitry of drug reward. The starting point for the reward circuit is the medial forebrain bundle, composed of myelinated fibers that bidirectionally connect the olfactory tubercle and nucleus accumbens with the hypothalamus and ventral tegmental area (
19), and includes ascending monoamine pathways such as the mesocorticolimbic dopamine system (
14). The initial action of drug reward is hypothesized to depend on dopamine release in the nucleus accumbens for cocaine, amphetamine, and nicotine, opioid peptide receptor activation in the ventral tegmental area (via dopamine activation) and nucleus accumbens (independent of dopamine activation) for opiates, and GABA
A systems in the nucleus accumbens and amygdala for alcohol. The nucleus accumbens is situated strategically to receive important limbic information from the amygdala, frontal cortex, and hippocampus that could be converted to motivational action via its connections with the extrapyramidal motor system. Thus, an early critical role of the nucleus accumbens was established for the acute reinforcing effects of drugs, with a supporting role of the central nucleus of the amygdala and ventral pallidum.
NEUROBIOLOGICAL MECHANISMS OF THE WITHDRAWAL/NEGATIVE AFFECT STAGE
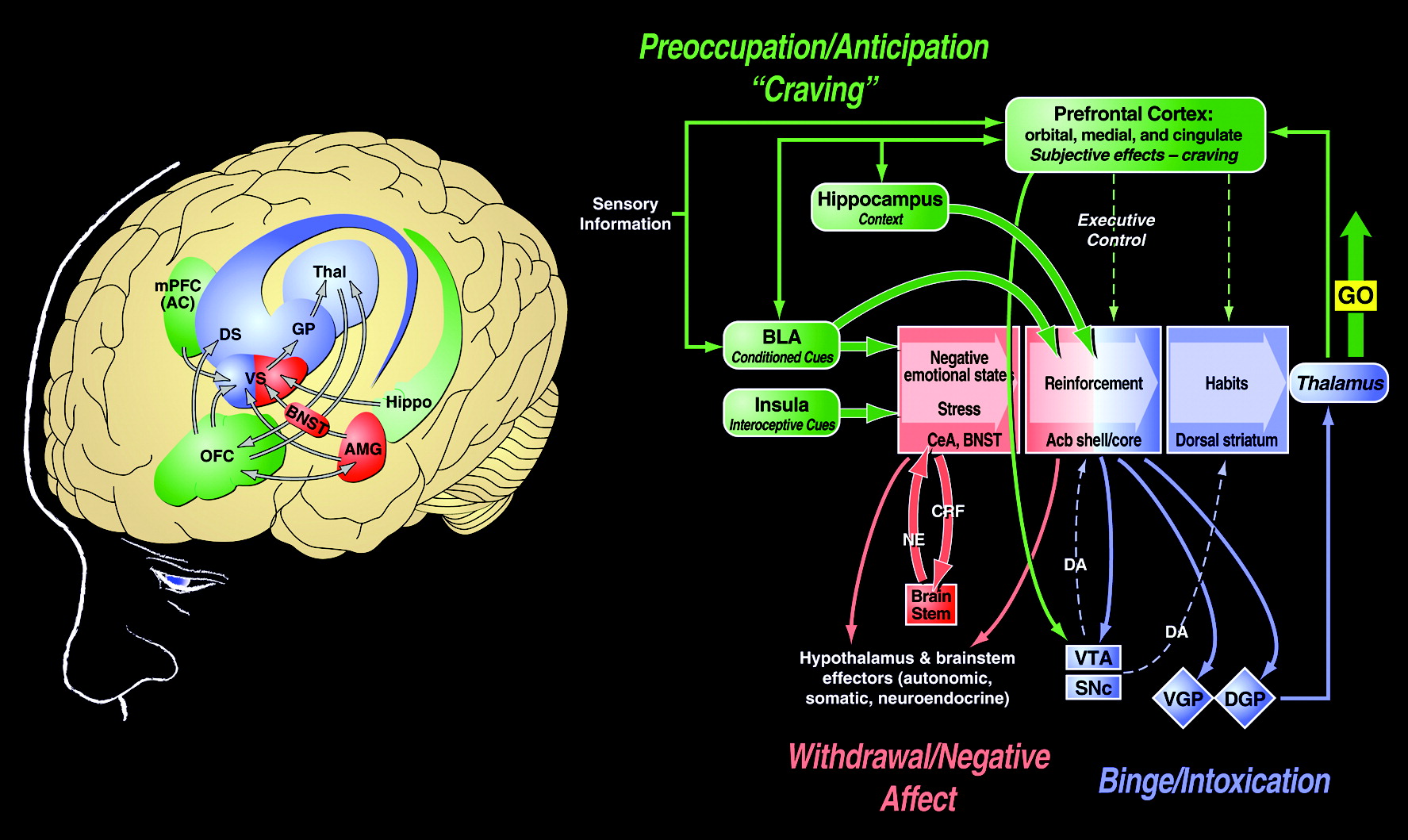
The neuroanatomical entity termed the extended amygdala (
20) may represent a common anatomical substrate that integrates brain arousal-stress systems with hedonic processing systems to produce the negative emotional states that drive negative reinforcement mechanisms associated with the development of addiction. The extended amygdala is composed of the bed nucleus of the stria terminalis, the central nucleus of the amygdala, and a transition zone in the medial subregion of the nucleus accumbens (shell of the nucleus accumbens). Each of these regions has cytoarchitectural and circuitry similarities (
20). The extended amygdala receives numerous afferents from limbic structures, such as the basolateral amygdala and hippocampus, and sends efferents to the medial part of the ventral pallidum and a large projection to the lateral hypothalamus, thus further defining the specific brain areas that interface classical limbic (emotional) structures with the extrapyramidal motor system (
21). The extended amygdala has long been hypothesized to play a key role not only in fear conditioning (
22) but also in the emotional component of pain processing (
23).
The neural substrates and neuropharmacological mechanisms for the negative motivational effects of drug withdrawal may involve disruption of the same neural systems implicated in the positive reinforcing effects of drugs but also involve recruitment of antireward systems (see below). Measures of brain reward function during acute abstinence from all major drugs with dependence potential have revealed increases in brain reward thresholds measured by direct brain stimulation reward (
24–
29). These increases in reward thresholds may reflect decreases in the activity of reward neurotransmitter systems in the midbrain and forebrain implicated in the positive reinforcing effects of drugs.
Changes at the neurochemical level that reflect changes in the neurotransmitter system implicated in acute drug reward have been hypothesized to reflect a within-system neuroadaptation and contribute significantly to the negative motivational state associated with acute drug abstinence. A within-system neuroadaptation can be defined as “the primary cellular response element to the drug would itself adapt to neutralize the drug's effects; persistence of the opposing effects after the drug disappears would produce the withdrawal response” (
30). Such within-system changes include decreases in dopaminergic transmission in the nucleus accumbens during drug withdrawal measured by in vivo microdialysis (
31,
32), increased sensitivity of opioid receptor transduction mechanisms in the nucleus accumbens during opioid withdrawal (
33), decreased GABA-ergic and increased
N-methyl-D-aspartate (NMDA) glutamatergic transmission during alcohol withdrawal (
34–
37), and differential regional changes in nicotinic receptor function (
38,
39). The decreased reward system function may persist in the form of long-term biochemical changes that contribute to the clinical syndrome of protracted abstinence and vulnerability to relapse.
The emotional dysregulation associated with the
withdrawal/negative affect stage also may involve a between-system neuroadaptation, in which neurochemical systems other than those involved in the positive rewarding effects of drugs of abuse are recruited or dysregulated by chronic activation of the reward system (
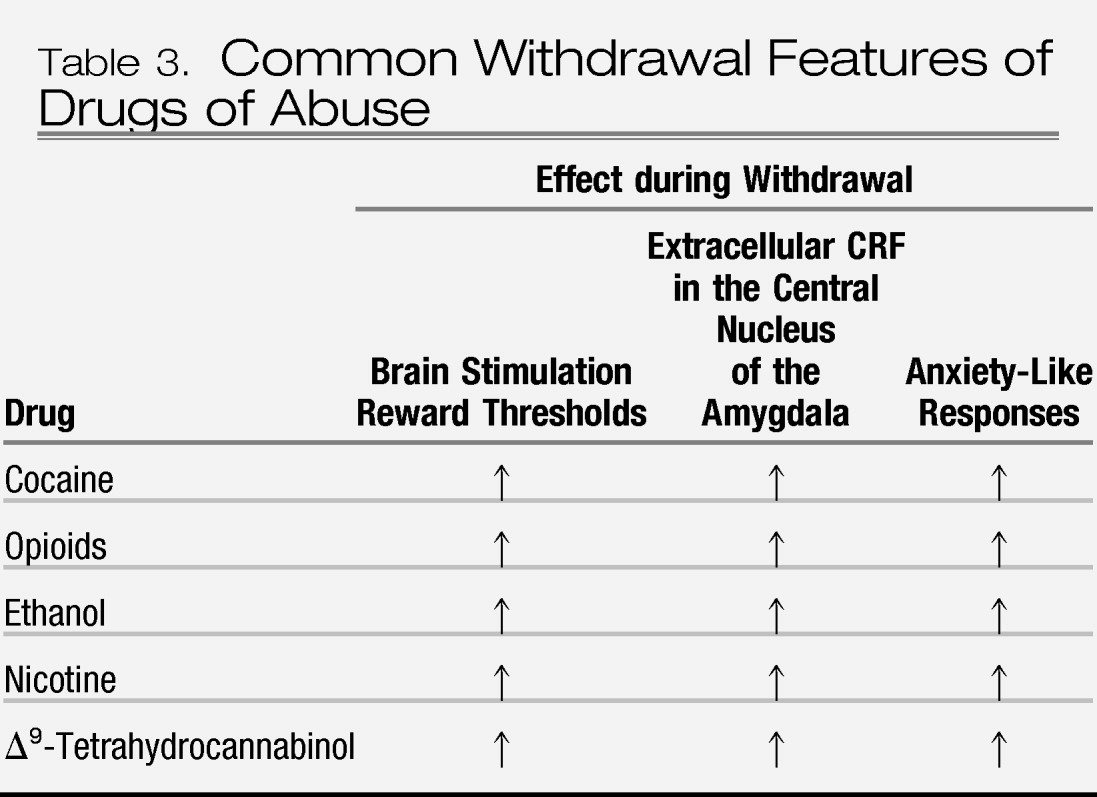
30). Brain neurochemical systems involved in stress modulation may also be engaged within the neurocircuitry of the brain stress systems in an attempt to overcome the chronic presence of the perturbing drug and to restore normal function despite the presence of drug. Both the hypothalamic-pituitary-adrenal axis and the brain stress system mediated by corticotropin-releasing factor (CRF) are dysregulated by chronic administration of all major drugs with dependence or abuse potential, with a common response of elevated ACTH, corticosterone, and amygdala CRF during acute withdrawal (
40–
45) (
Table 3). Acute withdrawal from drugs may also increase the release of norepinephrine in the bed nucleus of the stria terminalis and decrease levels of neuropeptide Y in the central and medial nuclei of the amygdala (
46).
These results suggest, during the development of dependence, not only a change in the function of neurotransmitters associated with the acute reinforcing effects of drugs (dopamine, opioid peptides, serotonin, GABA, and endocannabinoids) but also recruitment of the brain stress system (CRF and norepinephrine) and dysregulation of the neuropeptide Y brain antistress system (
5) (
Figure 3). Additionally, activation of the brain stress systems may not only contribute to the negative motivational state associated with acute abstinence but also may contribute to the vulnerability to stressors observed during protracted abstinence in humans.
Another candidate for the aversive effects of drug withdrawal is the opioid peptide dynorphin. Much evidence shows that dynorphin is increased in the nucleus accumbens in response to dopaminergic activation and, in turn, that overactivity of dynorphin systems can decrease dopaminergic function. κ opioid agonists are aversive, and cocaine, opioid, and ethanol withdrawal is associated with increased dynorphin in the nucleus accumbens or amygdala. Dynorphin systems may also interact with the brain CRF systems and evidence shows that dynorphin drives CRF and CRF drives dynorphin (
47).
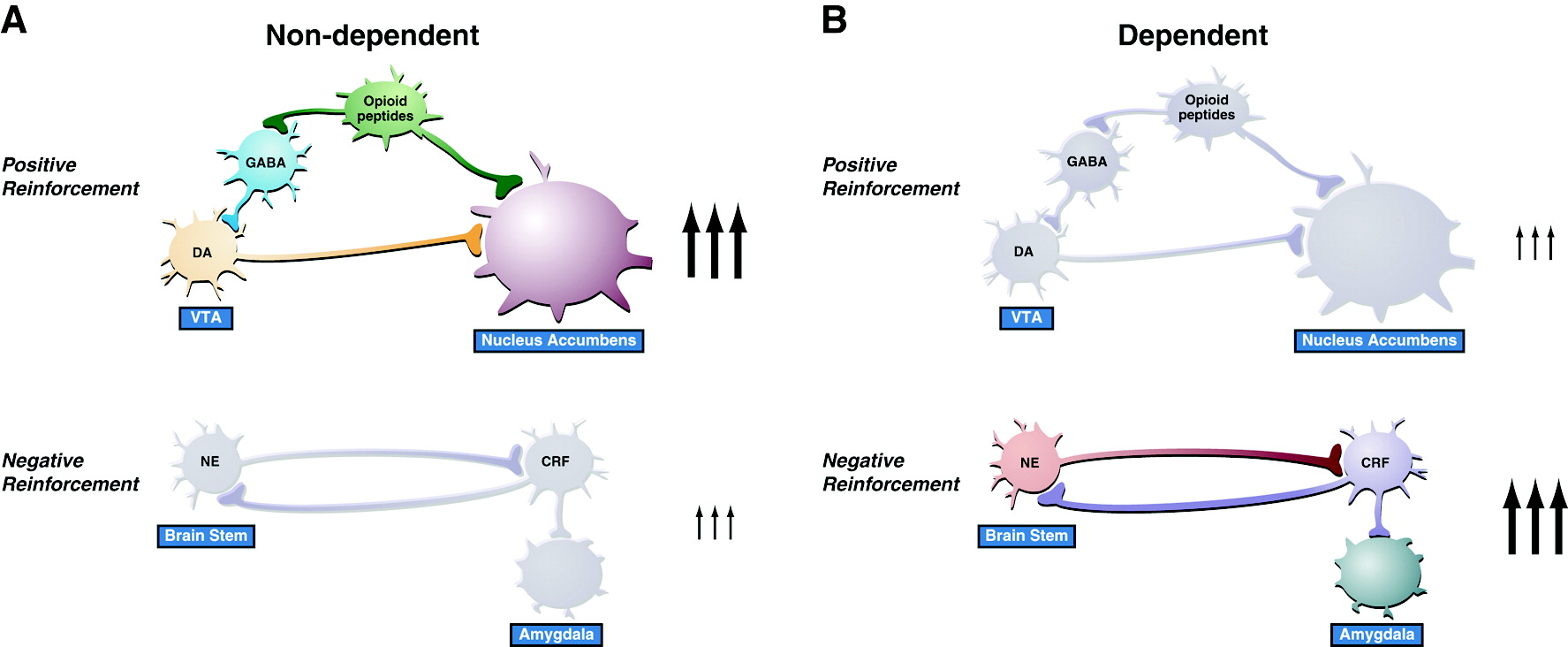
The concept of an antireward system has been formulated to accommodate the significant changes in brain emotional systems associated with the development of dependence (
48). The antireward concept is based on the hypothesis that there are brain systems in place to limit reward (
30), an opponent process concept that forms a general feature of biological systems. The concept of an antireward system is derived from the hypothesis of between-system neuroadaptations to activation of the reward system at the neurocircuitry level. A between-system neuroadaptation is a circuitry change in which circuit B (antireward circuit) is activated by circuit A (reward circuit) (
Figure 4). This concept has its origins in the theoretical pharmacology that predates opponent process theory (
49). Thus, the activation of brain stress systems such as CRF, norepinephrine, and dynorphin with concomitant dysregulation of the neuropeptide Y system may represent the recruitment of an antireward system in the extended amygdala that produces the motivational components of drug withdrawal and provides a baseline hedonic shift that facilitates craving mechanisms (
48).
NEUROBIOLOGICAL MECHANISMS OF THE PREOCCUPATION/ANTICIPATION STAGE
The
preoccupation/anticipation stage of the addiction cycle has long been hypothesized to be a key element of relapse in humans and defines addiction as a chronic relapsing disorder. Although often linked to the construct of craving, craving per se has been difficult to measure in human clinical studies (
50) and often does not correlate with relapse. Nevertheless, the stage of the addiction cycle in which the individual reinstates drug-seeking behavior after abstinence remains a challenging focus for neurobiological mechanisms and medications development for treatment.
Animal models of craving can be divided into two domains: drug seeking induced by stimuli paired with drug-taking and drug seeking induced by an acute stressor or a state of stress (
Table 4). Craving Type-1 animal models involve the use of drug-primed reinstatement and cue-induced reinstatement. Craving Type-2 animal models involve stress-induced reinstatement in animals that have acquired drug self-administration and then have been subjected to extinction of responding for the drug (
51).
Most evidence from animal studies suggests that drug-induced reinstatement is localized to the medial prefrontal cortex/nucleus accumbens/ventral pallidum circuit mediated by the neurotransmitter glutamate (
52). In contrast, neuropharmacological and neurobiological studies using animal models for cue-induced reinstatement involve the basolateral amygdala as a critical substrate with a possible feed-forward mechanism through the prefrontal cortex system involved in drug-induced reinstatement (
53,
54). Neurotransmitter systems involved in drug-induced reinstatement involve a glutamatergic projection from the frontal cortex to the nucleus accumbens that is modulated by dopamine activity in the frontal cortex. Cue-induced reinstatement involves dopamine modulation in the basolateral amygdala and a glutamatergic projection to the nucleus accumbens from both the basolateral amygdala and ventral subiculum (
53,
55). In contrast, stress-induced reinstatement of drug-related responding in animal models appears to depend on the activation of both CRF and norepinephrine in elements of the extended amygdala (central nucleus of the amygdala and bed nucleus of the stria terminalis) (
56,
57). Protracted abstinence, largely described in alcohol dependence models, appears to involve overactive glutamatergic and CRF systems (
58,
59).
In humans, cognitive deficits with addiction are observed that reflect the function of the medial prefrontal cortex, orbitofrontal cortex, and hippocampus (
60). Human subjects with cocaine addiction show impaired performance in tasks that involve attention, cognitive flexibility, and delayed reward discounting that are mediated by the medial and orbital prefrontal cortex, as well as spatial, verbal, and recognition memory impairments that are mediated by the hippocampus, and these deficits can predict poor treatment outcomes (
61,
62). Parallel animal studies of the orbitofrontal cortex, prefrontal cortex, and hippocampus in addiction have begun to show some of the deficits reflected in human studies. Experimenter-administered cocaine produced impairments in reversal learning (an orbitofrontal task) in rats and monkeys (
63–
65). Perhaps even more compelling, animals allowed extended access, but not limited access, to cocaine showed deficits in working memory (a prefrontal cortex-dependent task), a sustained attention task (a prefrontal cortex-dependent task), and an objection recognition task (a hippocampus-dependent task) (
66–
68).
MOLECULAR AND CELLULAR TARGETS WITHIN THE BRAIN CIRCUITS ASSOCIATED WITH ADDICTION
The focus of the present review is on the neuroplasticity of addiction that involves alterations in specific neurochemical systems in the context of the three stages of the addiction cycle. However, parallel to the neuroplasticity of the neurocircuitry are the molecular changes that occur in these same structures. Chronic exposure to opiates and cocaine leads to activation of cyclic adenosine monophosphate response-element binding protein (CREB) in the nucleus accumbens and central nucleus of the amygdala (
69,
70). CREB can be phosphorylated by protein kinase A and by protein kinase regulated by growth factors, putting it at a point of convergence for several intracellular messenger pathways that can regulate gene expression. Activation of CREB in the nucleus accumbens with psychostimulant drugs is linked to the motivational symptoms of psychostimulant withdrawal, such as dysphoria, possibly through the induction of the opioid peptide dynorphin, which binds to κ opioid receptors and has been hypothesized to represent a mechanism of motivational tolerance and dependence (
15). Repeated CREB activation drives dynorphin expression in the nucleus accumbens, which in turn decreases dopaminergic activity and may activate other brain stress systems, all of which can contribute to negative emotional states. Extracellular signal-regulated kinase (ERK) is another key element of intracellular signaling that is considered a key component in the plasticity associated with repeated administration of cocaine, specifically behavioral sensitization, cocaine reward, and time-dependent increases in cocaine seeking after withdrawal (i.e., incubation effect) (
71,
72).
Another molecular target for regulating the plasticity that leads to addiction is dysregulation of cystine-glutamate exchange, which is hypothesized to drive pathological glutamate signaling related to several components of the addiction cycle. Repeated administration of cocaine blunts cystine-glutamate exchange, leading to reduced basal and increased cocaine-induced glutamate in the nucleus accumbens that persists for at least 3 weeks after the last cocaine treatment (
73). Most compelling is the observation that treatment with
N-acetylcysteine, by activating cystine-glutamate exchange, prevented cocaine-induced escalation and behavioral sensitization, restored the ability to induce long-term potentiation and long-term depression in the nucleus accumbens, and blunted reinstatement in animals and conditioned reactivity to drug cues in humans (
74–
76).
Finally, CREB and other intracellular messengers can activate transcription factors, which can change gene expression and produce long-term changes in protein expression, and, as a result, neuronal function. Although acute administration of drugs of abuse can cause rapid (within hours) activation of members of the Fos protein family, such as c-
fos, FosB, Fra-1, and Fra-2 in the nucleus accumbens, other transcription factors and isoforms of ΔFosB (i.e., a highly stable form of FosB) have been shown to accumulate over longer periods of time (days) with repeated drug administration
(15). Animals with activated ΔFosB have exaggerated sensitivity to the rewarding effects of drugs of abuse, and ΔFosB has been hypothesized to act as a sustained molecular “switch” that helps initiate and maintain a state of addiction (
77). Whether (and how) such transcription factors influence the function of the brain stress systems, such as CRF, dynorphin, neuropeptide Y, and the others described above, remains to be further explored.
BRAIN IMAGING CIRCUITS INVOLVED IN HUMAN ADDICTION
Brain imaging studies using positron emission tomography with ligands for measuring oxygen utilization or glucose metabolism or using MRI techniques are providing dramatic insights into the neurocircuitry changes in the human brain associated with the development, maintenance, and vulnerability to addiction. These imaging results overall show a striking resemblance to the neurocircuitry identified by human studies. During acute intoxication with alcohol, nicotine, and cocaine, there is an activation of the orbitofrontal cortex, prefrontal cortex, anterior cingulate, extended amygdala, and ventral striatum. This activation is often accompanied by an increase in availability of the neurotransmitter dopamine. During acute and chronic withdrawal, a reversal of these changes occurs, with decreases in metabolic activity, particularly in the orbitofrontal cortex, prefrontal cortex, and anterior cingulate, and decreases in basal dopamine activity as measured by decreased D
2 receptors in the ventral striatum and prefrontal cortex. Cue-induced reinstatement appears to involve the reactivation of these circuits, much like acute intoxication (
78–
80). Craving or cues associated with cocaine and nicotine activated the prefrontal cortex and anterior cingulate gyrus (
81,
82). Imaging studies also show evidence that cues associated with cocaine craving increase dopamine release in the striatum as well as opioid peptides in the anterior cingulate and frontal cortex (
83–
85). Craving in alcoholics appears to be correlated with higher opioid peptide activity in the striatum but lower dopaminergic activity (
86,
87). Thus, imaging studies reveal baseline decreases in orbitofrontal function and dopamine function during dependence but the reactivation of dopamine and reward system function during acute craving episodes, consistent with the early formulation of different neural substrates for craving Type-1 and Type-2 (see above).
SUMMARY AND CONCLUSIONS
Much progress in neurobiology has provided a useful neurocircuitry framework with which to identify the neurobiological and neuroadaptive mechanisms involved in the development of drug addiction. The brain reward system implicated in the development of addiction is composed of key elements of the basal forebrain with a focus on the nucleus accumbens and central nucleus of the amygdala. Neuropharmacological studies in animal models of addiction have provided evidence for the activation of specific neurochemical mechanisms in specific brain reward neurochemical systems in the basal forebrain (dopamine, opioid peptides, GABA, and endocannabinoids) during the binge/intoxication stage. During the withdrawal/negative affect stage, dysregulation of the same brain reward neurochemical systems occurs in the basal forebrain (dopamine, opioid peptides, GABA, and endocannabinoids). There is also recruitment of brain stress/aversion systems (CRF and dynorphin) and dysregulation of brain antistress systems (neuropeptide Y) that contribute to the negative motivational state associated with drug abstinence. During the preoccupation/anticipation stage, neurobiological circuits that engage the frontal cortex glutamatergic projections to the nucleus accumbens are critical for drug-induced reinstatement, whereas basolateral amygdala and ventral subiculum glutamatergic projections to the nucleus accumbens are involved in cue-induced reinstatement. Stress-induced reinstatement appears to be mediated by changes in the antireward systems of the extended amygdala. The changes in craving and antireward (stress) systems are hypothesized to remain outside of a homeostatic state, and as such convey the vulnerability for the development of dependence and relapse in addiction. Genetic studies to date in animals suggest roles for the genes encoding the neurochemical elements involved in the brain reward (dopamine, opioid peptide) and stress (neuropeptide Y) systems in the vulnerability to addiction. Molecular studies have identified transduction and transcription factors that may mediate the dependence-induced reward dysregulation (CREB) and chronic-vulnerability changes (ΔFosB) in neurocircuitry associated with the development and maintenance of addiction. Human imaging studies reveal similar neurocircuits involved in acute intoxication, chronic drug dependence, and vulnerability to relapse.
Although no exact imaging results necessarily predict addiction, two salient changes in established and unrecovered substance-dependent individuals that cut across different drugs are decreases in orbitofrontal/prefrontal cortex function and decreases in brain dopamine D2 receptors. No molecular markers are sufficiently specific to predict the vulnerability to addiction, but changes in certain intermediate early genes with chronic drug exposure in animal models show promise of long-term changes in specific brain regions that may be common to all drugs of abuse. The continually evolving knowledge base of the biological and neurobiological aspects of substance use disorders provides a heuristic framework to better develop diagnoses, prevention, and treatment of substance abuse disorders.