INTRODUCTION
Anxiety disorders are marked by excessive fear (and avoidance), often in response to specific objects or situations and in the absence of true danger, and they are extremely common in the general population. According to a recent epidemiological study, the lifetime prevalence of any anxiety disorder is 28.8% (
Kessler et al, 2005). Anxiety disorders are associated with impaired workplace performance and hefty economic costs (
Greenberg et al, 1999), as well as an increased risk of cardiovascular morbidity and mortality (
Albert et al, 2005;
Kawachi et al, 1994;
Smoller et al, 2007). Given that anxiety disorders are a significant problem in the community, recent neuroimaging research has focused on determining the brain circuits that underlie them to inform the use of existing treatments and guide the possible development of new treatments. In the future, neuroimaging studies of anxiety disorders may also prove to be clinically helpful in the prediction of treatment response.
Given that excessive fear is a key component of anxiety disorders, it is not surprising that the search for the neurocircuitry of anxiety disorders has its roots in and has been closely intertwined with studies of fear circuits in animal models. A large volume of experimental work has examined the neurocircuitry associated with fear responses, mainly in rodents, using primarily fear conditioning, inhibitory avoidance, and fear-potentiated startle models. Key components of fear circuitry including the amygdala (and its subnuclei), nucleus accumbens (including bed nucleus of stria terminalis BNST), hippocampus, ventro-medial hypothalamus, periaqueductal gray, a number of brain stem nuclei, thalamic nuclei, insular cortex, and some prefrontal regions (mainly infralimbic cortex) have been identified in these studies (for recent reviews see
Davis, 2006;
Maren, 2008;
Quirk and Mueller, 2008). These regions have their respective roles in the various components of fear processing such as the perception of threat or of unconditioned stimuli, the pairing of an unconditioned stimulus and conditioned response (learning/conditioning), the execution of efferent components of fear response, and the modulation of fear responses through potentiation, contextual modulation, or extinction. Some key findings from animal literature, such as the central role of amygdaloid nuclei in the acquisition of fear conditioning and expression of fear responses, the involvement of the hippocampus in contextual processing, and the importance of the infralimbic cortex in extinction recall, have been replicated across different studies and laboratories. These basic components of fear circuitry are well preserved across species and likely support similar functions in humans. Animal work using
in vivo electrophysiological recording, tracing and lesions/reversible inactivation techniques was indispensable in acquiring this knowledge. Some recent work had even suggested that there might be separate fear and anxiety systems orchestrated through the central nucleus of the amygdala and the BNST, respectively (
Davis, 2006). These types of findings are particularly exciting as they might allow for a better focus on the neurocircuits involved in pathological anxiety.
On the other hand, other important issues, such as the exact neuroanatomical region that stores fear memory traces, or the exact role of a particular process (eg, the role of reconsolidation in fear memory,
Nader and Hardt, 2009), or of a particular region (eg, the insular cortex) are intensely debated and actively studied. Nevertheless, the basic fear-related neurocircuitry identified in rodents is a useful place to start examining anxiety-related neurocircuitry in humans. It is important to note that the exact roles of many brain regions are yet to be firmly established and could differ across species. Even regions such as the amygdala, hippocampus, and nucleus accumbens might be involved in different, additional, or even unique tasks in humans (eg, the role of the hippocampus in explicit verbal memory in humans). Finally, there are major differences between human anxiety/anxiety disorders and fear conditioning models in animals. These differences include the frequent absence of clear unconditioned stimuli (US) in human anxiety disorders, and the central roles of avoidance and cognitive components (eg, anticipatory anxiety) in humans. These unique characteristics of anxiety disorders suggest potential involvement of other brain regions in addition to those identified in rodents, such as areas of prefrontal cortex that are more unique to humans. Thus, although animal studies are indispensable in understanding basic fear neurocircuitry,
in vivo human studies are critical for understanding the neurocircuitry of anxiety disorders.
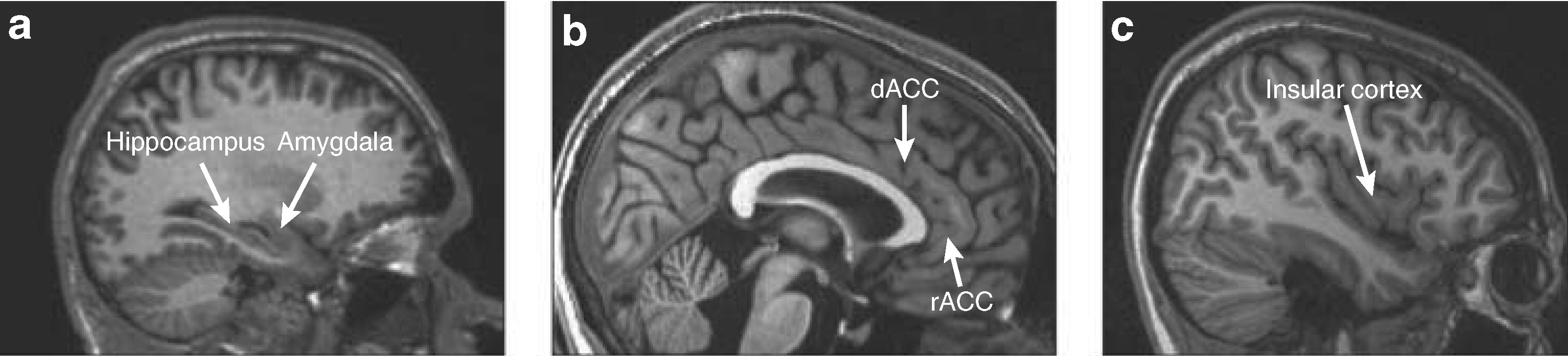
In this review, we will discuss three main topics: (1) fear neurocircuitry in healthy humans; (2) stress as a normal response to internal and external stimuli, and (3) anxiety disorders as defined in human psychopathology. The first of these topics will include a discussion of Pavlovian fear conditioning and extinction, pharmacologically induced fear and anxiety states, and the assessment of emotional stimuli in humans. In the third topic, we review the role of brain regions such as the amygdala, medial prefrontal cortex (including the rostral anterior cingulate cortex (rACC) and dorsal anterior cingulate cortex (dACC)), hippocampus, and insular cortex in anxiety disorders (
Figure 1). Finally, we discuss some of the limitations of neuroimaging studies of anxiety disorders as well as the directions that we expect the field to take in the near future.
STRESS
An important and often overlooked aspect of the fear/anxiety neurocircuitry is its overlap and interaction with the neurocircuitry that orchestrates the stress response. It is important to note that the concept of ‘stress’ used here is relatively specific. It does not encompass general concepts of ‘subjective distress’ or ‘performance load.’ Although these are useful concepts, they are heterogeneous by nature and are not likely to be associated with a specific neurocircuitry. On the other hand, the concept of a stress system that leads to activation of limbic-hypothalamopituitary-adrenal axis (LHPA) and secretion of stress hormones like corticotropin-releasing hormone (CRH), adrenocorticotropic hormone, and cortisol is quite specific and is likely to be highly relevant to the neurocircuitry of fear and anxiety. The neurocircuitry governing LHPA activation has been the focus of intense studies in rodents, primates, and humans because it has been repeatedly linked to the neurobiology of mood disorders (which is addressed in detail elsewhere in this volume), but the evidence linking LHPA axis abnormalities to anxiety disorders has been less consistent, sometimes confusing, and often oversimplified. At the same time, some of the same brain regions are implicated in both anxiety and stress responses, suggesting that these responses are interrelated and can influence each other. Furthermore, anxiety and mood disorders are highly comorbid, suggesting that some common abnormalities in neurocircuitry might be present in both disorders. In the following few paragraphs, we briefly address only the structural overlap in neurocircuits and the effects of stress system activation (or stress hormones) on anxiety/fear neurocircuitry.
Epidemiologically, major depression is highly comorbid with anxiety disorders like PTSD, panic disorder, and social phobia (
Reiger et al, 1990), and anxiety symptoms are highly prevalent in depression (
Frances et al, 1990). Furthermore, major subcortical components of the LHPA axis (eg, hypothalamus, hippocampus, amygdala, and BNST) have also been identified as key components of anxiety/fear neurocircuitry (albeit sometimes involving different subnuclei, for example paraventricular nuclei
vs ventromedial hypothalamus for LHPA and fear neurocircuitry, respectively). More recently with the introduction of
in vivo imaging methodologies in LHPA/stress research, the role of cortical structures like the insula and dorsal mPFC in the activation and inhibition of stress response, respectively, has been reported (
Liberzon and Martis, 2006) as well as the role of subgenual ACC in self-induced sadness and depression (
Mayberg et al, 1999). Together, these findings suggest a significant overlap in structures involved in the stress response and those involved in fear/anxiety responses (eg medial prefrontal cortex, insula, amygdala, hippocampus, and BNST). Finally, with respect to neurotransmitters involved, CRH is likely involved in the orchestration of both LHPA axis activity and many anxiety/fear responses. (For a review see
Heim and Nemeroff, 2001.)
The activation of these overlapping regions in functional neuroimaging studies does not necessarily signify, however, activation of both the fear/anxiety response and the LHPA axis. As a matter of fact, activation of fear/anxiety does not necessarily activate an LHPA stress response, even in highly fearful (phobic) individuals (
Curtis et al, 1976). In turn, activation of LHPA axis is not necessarily experienced subjectively as fear or anxiety. For example, morning awakening, food intake, and nausea all lead to LHPA axis activation without notable increases in subjective sense of fear. It is becoming increasingly clear that specific characteristics of experience (novelty, control, social support, etc.) are more salient for LHPA axis activation than degree of subjective distress or fear (
Abelson et al, 2007). These facts help to better understand the findings of non-specific, or even sometimes counterintuitive, findings regarding the LHPA and stress responses in anxiety disorders such as panic disorder (
Abelson et al, 2007) and PTSD (
Yehuda, 2006;
Yehuda et al, 1991). This also suggests that activation in specific cortical regions like mPFC or insula cannot be readily interpreted as a component of the fear response, and has to be considered within a context of a specific experiment, subjective report, symptoms, neuroendocrine profile, etc.
With these caveats in mind, important findings about stress exposure and LHPA axis activation affecting fear/anxiety responses have been accumulating. These can be seen in two general categories: (1) the immediate effects of stress or of stress hormones on fear/anxiety responses (eg, stress or stress hormone exposure immediately precedes, or is present during the fear/anxiety responses), and (2) delayed or developmental effects, (eg, stress exposure during developmentally sensitive periods, like early childhood, modulates fear/anxiety responses later in life). In the former category, it has been reported that exposure to acute stress in healthy individuals potentiates the anxiety response (
Grillon et al, 2007). In addition, stress exposure (Trier Social Stress Test) led to enhanced galvanic skin responses to conditioned stimuli (CS+) during fear conditioning (
Jackson et al, 2006). Interestingly, stress modulates fear responses differentially in men and women. Differential effects of stress on fear/anxiety in females
vs males also have been demonstrated in animal studies. Chronic stress exposure led to impaired extinction recall of fear conditioning in male but not female rats (
Baran et al, 2009). Stress exposure in animal studies also led to enhancement in contextual fear conditioning (
Cordero et al, 2003). The effects of stress hormone exposure are somewhat more difficult to interpret because higher endogenous cortisol levels were associated with higher skin conductance responses (SCR) (
Jackson et al, 2006), whereas administration of exogenous cortisol led to decreased SCR (
Stark et al, 2006).
With respect to delayed effects of stress during the vulnerable developmental period, the findings are somewhat complex. Studies of early maternal separation in rodents (
Plotsky and Meaney, 1993) and variable foraging in primates (
Coplan et al, 1996) have revealed long-term alteration in stress axis responses and key neurotransmitter systems (for review see
Heim and Nemeroff, 2001). In addition, recent findings of gene-by-environment interactions in PTSD (
Binder et al, 2008) also point toward the possibility that early childhood experience might modify fear/anxiety neurocircuitry and contribute to the development of anxiety disorders. Direct evidence of these effects on fear/anxiety behavior is less convincing, however, as maternal separation in rat pups, which did alter relevant neurotransmitter systems, did not result in significantly enhanced startle response or decreased open field exploration as compared with non-separated animals (
Caldji et al, 2000). Similarly, mixed results have been obtained in other studies where rats exposed to severe sporadic stress spent more time in open arms of an elevated plus maze but displayed increases in defensive probe burying behavior. Furthermore, animals exposed to milder chronic stress showed opposite changes (
Pohl et al, 2007).
The character of the stress exposure (mild
vs severe, prolonged
vs short, predictable
vs non-predictable) and the sex of the individual emerge as important variables that can define the long-term effects of stress exposure, but more experimental data are clearly needed. Similarly, little is known about the specific mechanisms by which stress exposure modulates fear/anxiety circuitry. It has been suggested, as stated above, that early developmental stress exposure alters fear/anxiety circuitry via altered sensitivity and responsivity of the CRH and adrenergic systems, and recent advances in morphological work had suggested a potential mechanism for the effects of stress on fear conditioning and extinction. Chronic stress decreases dendritic branching in the hippocampus (
McEwen, 2001) and mPFC (
Liston et al, 2006;
Radley et al, 2004), but increases dendritic branching in the amygdala (
Mitra et al, 2005;
Vyas et al, 2006). This pattern could lead to increased conditioning and impaired extinction, and both of these processes could contribute changes in anxiety/fear-related behaviors. Future research addressing these important questions will be needed to fully understand the impact of stress/LHPA axis activation on fear/anxiety and the underlying neurocircuitry.
Summary
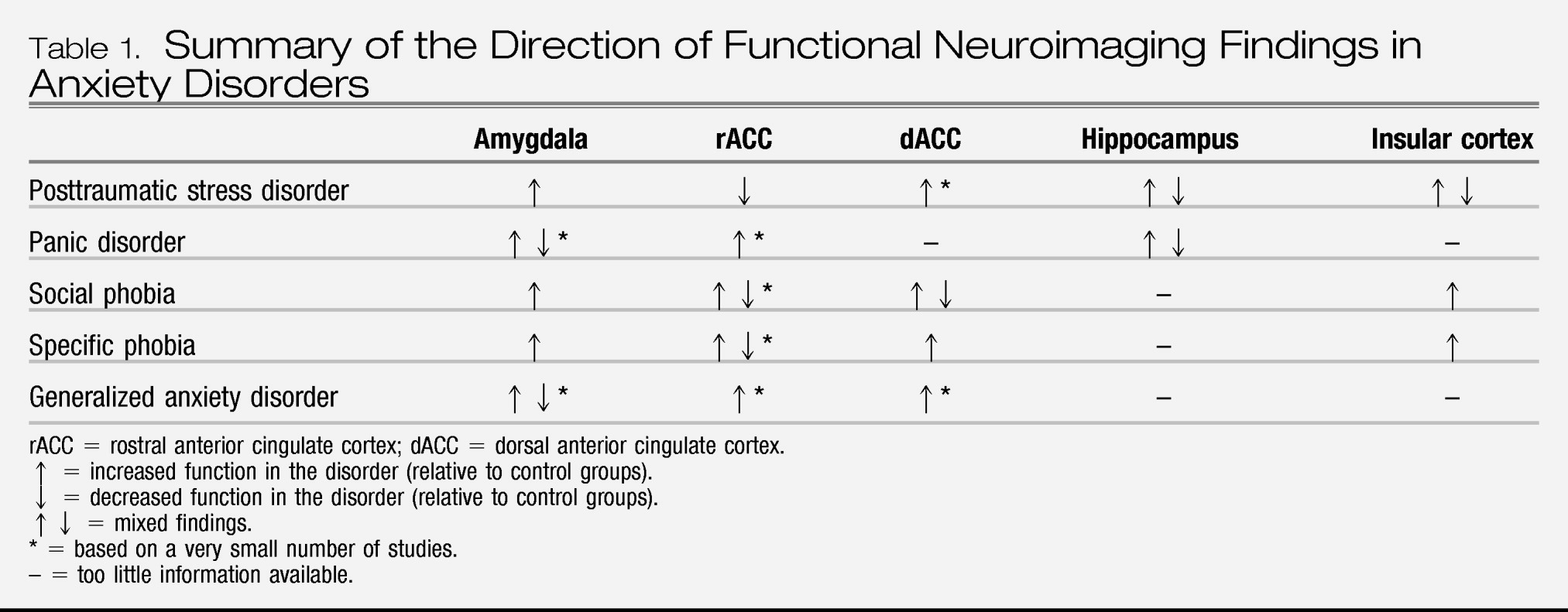
Overall, the findings of functional neuroimaging studies are consistent with the notion of exaggerated amygdala activation to specific stimuli in a number of anxiety disorders, especially social phobia, specific phobia, and PTSD. Facial expressions have been especially effective probes of amygdala responses in social phobia and PTSD. Interestingly, the data regarding amygdala function in panic disorder are still inconclusive, and given that relatively few studies have examined amygdala function in GAD, additional research is needed to make meaningful comparisons of amygdala responses between various anxiety disorders. Although increased amygdala activation has been observed in a few studies of OCD, the overall pathophysiology of OCD appears to be localized to a different brain circuit. One potential conceptualization of these findings is that amygdala hyperactivation is a common pathway for exaggerated anxiety/fear that is triggered by specific stimuli. Thus anxiety disorders that manifest increased fear/anxiety that is associated with specific identifiable stimuli (eg, PTSD, social phobia, and specific phobia) will have evidence of exaggerated amygdala reactivity. Panic attacks, on the other hand, can occur in the absence of such stimuli and might thus involve activation of other structures within fear/anxiety neurocircuitry (such as brain stem nuclei, periaqueductal gray, and mPFC). If this is the case, it can explain why the degree of amygdala activation in panic disorder may vary depending on the presence of identified panic-related stimuli and/or other brain regions activated. Conceptually, the hyperresponsivity of amygdala and brain stem is consistent with cognitive and somatic manifestation of panic attacks; however, hyperactivation of the anterior cingulate is probably more consistent with compensatory/regulatory roles rather than reflecting panic-specific pathophysiology.
Consistent with the findings of the meta-analysis of
Etkin and Wager (2007), relatively diminished rACC activation has been reported fairly consistently in PTSD, but not in other anxiety disorders. Thus, relatively diminished rACC function may be specific to PTSD and could reflect abnormalities in recall/contextualization of fear memories. Activation in other regions like the dACC and insular cortex appears to be elevated in PTSD and in several of the other anxiety disorders. Exaggerated activation in these regions may reflect various aspects of anxiety/fear response, such as anticipatory anxiety, interoceptive components, autobiographic memory, or anxiety proneness (
Paulus and Stein, 2006;
Shin et al, in press;
Simmons et al, 2006). Together these findings can be conceptualized as an evidence of hyperresponsive threat detection, autobiographic memory, and somatic/physiological reactivity systems in PTSD, accompanied by the failure in regulatory regions responsible for safety signaling, fear extinction, and stimulus appraisal, together leading to aberrant contextual processing of the threat-related stimuli.
The hippocampus was studied most frequently in PTSD and panic disorder, but rarely in the other disorders. Thus, whether similar structural and functional abnormalities in the hippocampus occur in other anxiety disorders is uncertain. Although the evidence for hippocampal involvement in memory and contextual processing is strongly supported by animal studies, and deficits in hippocampal function (eg, contextual processing) are consistent with the PTSD model outlined above, more information is needed to understand the role of the hippocampus in anxiety disorders.
However, neuroimaging studies of anxiety disorders are not without limitations. On the technical side, the spatial resolution of functional neuroimaging techniques is limited, and even those techniques with the best spatial resolution (fMRI) cannot accurately differentiate between very small adjacent structures (such as subnuclei of the amygdala). This, in addition to the presence of susceptibility artifacts (in the case of fMRI studies), makes resolution of potentially important structures, such as the hypothalamus and brain stem nuclei, particularly problematic for studies of fear/anxiety neurocircuitry. In addition, the temporal resolution of some of the imaging techniques used (ie, PET and SPECT) makes it difficult to detect quick and transient responses to stimuli. On the clinical side, the use of medications (ie, antidepressants or benzodiazepines) in patient groups can affect brain activation and represent a confounding factor. However, for regions such as the amygdala, medications tend to normalize amygdala responses rather than exaggerate them (
Harmer et al, 2006;
Paulus et al, 2005). Comorbidity represents a challenge as well. The majority of patients with anxiety disorders have comorbid conditions, such as depression. Although it is tempting to exclude from neuroimaging studies those anxiety disorder patients with comorbid conditions, this procedure raises concerns about the ability to generalize findings to the larger population of individuals with anxiety disorders. Recent studies have attempted to deal with this issue by separating anxiety disorder patients into groups with and without comorbidity (
Kemp et al, 2007;
Lanius et al, 2007).
FUTURE RESEARCH DIRECTIONS
Although much information that is relevant to the pathophysiology of anxiety disorders has been gained over the last two decades, many research questions remain to be answered. As we have noted previously, questions remain regarding the specific roles of the amygdala, medial prefrontal cortex, insula, and hippocampus in the anxiety disorders. In addition, one of the more general and basic questions is whether the functional abnormalities identified in anxiety disorders represent acquired signs of the disorders or vulnerability factors that increase the risk of developing the disorders. For example, does amygdala hyperresponsivity occur after the symptoms of social phobia, specific phobia, or PTSD appear? Or does the amygdala hyperresponsivity precede the development of symptoms and increase the risk for developing them? Two types of studies could be used to try to address these questions. First, longitudinal studies that include functional neuroimaging could assess functional activation before the onset of anxiety symptoms (or before trauma exposure in the case of PTSD) to determine whether baseline amygdala activation predicts (or increases the risk for) subsequent anxiety disorder diagnoses. One recent study using a variant of this longitudinal design has yielded findings suggesting that amygdala activation may represent a vulnerability factor. In this study, amygdala activation in response to novel faces was studied in adults who were categorized in childhood as either behaviorally inhibited or uninhibited (
Schwartz et al, 2003). Behavioral inhibition in childhood is a known risk factor for the development of social anxiety later in life (
Biederman et al, 2001;
Schwartz et al, 1999). Amygdala activation was greater in the inhibited group as compared with the uninhibited group, and this finding remained even when inhibited subjects with social phobia were removed from the analyses. Although this study does not provide definitive proof that exaggerated amygdala activation is a vulnerability factor for the development of social phobia, it does lend some support to the idea.
The second type of study that can help determine whether functional abnormalities are acquired characteristics or vulnerability factors involves studying the identical twins of probands with
vs without the disorder in question. With regard to PTSD, Roger Pitman and colleagues have studied combat veterans with PTSD and their combat-unexposed identical co-twins without PTSD, as well as combat veterans who never had PTSD and their identical combat-unexposed co-twins without PTSD. Structural and functional abnormalities that are observed in both the individuals with PTSD and their identical trauma-unexposed co-twins likely represent familial vulnerability factors, whereas abnormalities that are observed only in the individuals with PTSD would be consistent with acquired characteristics. Using this type of design, diminished hippocampal volumes (
Gilbertson et al, 2002) and dACC hypermetabolism (
Shin et al, in press) appear to be familial vulnerability factors, whereas diminished gray matter volumes in the rACC appear to be acquired characteristics (
Kasai et al, 2008).
This twin design, however, is unable to determine whether familial vulnerability is attributable to genetic
vs environmental factors. Future studies will be needed to determine whether the functional abnormalities that appear to be familial vulnerability factors are associated with specific genotypes. For example, dACC hypermetabolism has been reported in healthy carriers of the short allele of the serotonin transporter polymorphism (
Graff-Guerrero et al, 2005). The prevalence of the short allele appears to be elevated in PTSD (
Lee et al, 2005), and the finding of dACC hypermetabolism in individuals with PTSD and their identical co-twins could be at least in part attributable to the presence of this short allele. Future studies will evaluate this possibility. Studies that examine links between specific genotypes and endophenotypes (eg, neuroimaging or neuroendocrine measures) that may predispose individuals to various anxiety disorders following environmental modulation (during development or in adulthood) are likely to prove very valuable in future research.
Much more progress is urgently needed in examining
in vivo neurochemistry in anxiety disorders. SPECT, PET, and MRS studies are needed to link functional neuroimaging data with cellular and molecular changes that might be driving these abnormalities. Finally, another major future direction would entail using functional neuroimaging to predict treatment response in patients with anxiety disorders. This type of research has recently begun, and some studies have reported that greater amygdala activation at a pre-treatment baseline predicts a less favorable response to (1) cognitive-behavioral therapy in PTSD (
Bryant et al, 2008a), and (2) venlafaxine in GAD (
Whalen et al, 2008). Future studies will be needed to determine whether specific pre-treatment functional neuroimaging profiles can distinguish between those who will respond to medication
vs cognitive-behavioral treatments. One ultimate goal of this research would be to determine whether functional neuroimaging measures can be used to guide treatment choice for individual patients.