Although multiple autoantibodies recognizing synaptic receptors have been described, recent work also implicates disruption of
trans-synaptic scaffolding systems in certain autoimmune encephalitides.
Trans-synaptic neuronal cell adhesion molecules are known to be crucial for proper synapse formation and adhesion, plasticity, and function.
26 In both developing and mature neurons, these molecules also serve to recruit and anchor pre- and postsynaptic proteins to appropriate synaptic localizations, allowing for normal synaptic transmission. In some instances, neuropsychiatric disorders such as autism and schizophrenia are postulated to result from genetic mutations in these neuronal cell-adhesion systems.
27,28 Recent discoveries now indicate that acquired autoimmune syndromes also target
trans-synaptic signals. Leucine-rich glioma-inactivated 1 (LGI1) is a secreted protein that interacts with presynaptic ADAM23 and postsynaptic ADAM22 to create a
trans-synaptic protein complex, which also includes potassium channels and AMPA-type glutamate receptors.
29,30 Mutations in LGI1 are known to cause autosomal-dominant partial epilepsy with auditory features,
31 a syndrome characterized by temporal lobe seizures with prominent auditory hallucinations (
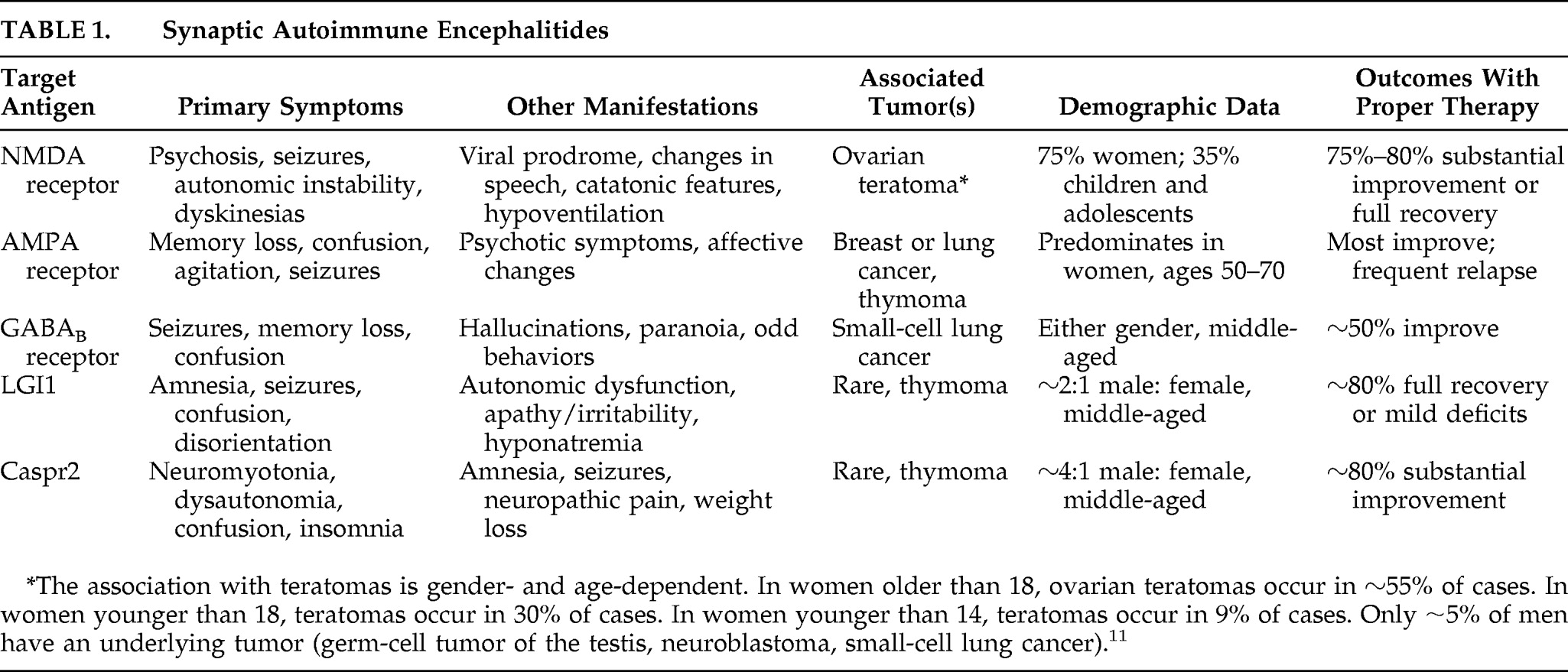
Table 1).
32 A classic limbic encephalitis previously thought to be caused by autoantibodies recognizing voltage-gated potassium channels (VGKC) is now known to result from autoantibodies targeting LGI1.
30,33 As described in detail as encephalitis attributed to anti-VGKC antibodies,
34 anti-LGI1 patients present most prominently with seizures, memory loss, and confusion. Other symptoms can include autonomic dysfunction (hyperhidrosis, hypersalivation) and behavioral changes such as apathy and irritability. MRI usually shows increased signal involving medial temporal lobes, although (uncharacteristic of classic limbic encephalitis) CSF is often normal. It is unclear why patients with anti-LGI1 antibodies do not often experience perceptual disturbances akin to those in patients with autosomal-dominant partial epilepsy with auditory features, although this difference is likely related to the acquired dysfunction of LGI1 later in life as opposed to a developmental abnormality. Like other autoimmune encephalopathies with extracellular antigen targets, anti-LGI1 encephalitis responds remarkably well to immunotherapy, with ∼80% of patients showing either full recovery or mild disability.
34,35In addition to LGI1, current research suggests that another molecule involved in neuronal cell adhesion might be a target for autoimmune syndromes.
30,33 Contactin-associated protein-like 2 (Caspr2) has a role in clustering VGKC at the paranodal regions of myelinated axons.
36 Caspr2 is a member of the neurexin superfamily, which mediates cell–cell interactions in the CNS and in which mutations have been associated with schizophrenia, autism, and mental retardation.
37 Genetic-analysis experiments have described a cortical dysplasia/focal epilepsy syndrome caused by mutations in Caspr2.
38 Patients with these mutations present early in life (between ages 2 and 7) with intractable seizures, followed by diminished learning and social behaviors, with language regression. Other pervasive neuropsychiatric symptoms are autistic-like and include hyperactivity, inattention, and aggression. Autoantibodies recognizing Caspr2 have been described in autoimmune encephalitis, often in association with symptoms of peripheral nerve hyperexcitabilty such as neuromyotonia (difficulty in muscle relaxation), cramps, fasciculations, and muscle spasms (
Table 1).
30,33,39 Taken together, these two autoimmune syndromes highlight the role of synaptic organizers in autoimmune encephalitis and open new avenues toward understanding the role of
trans-synaptic signals in disease states.