The clinical efficacy of typical neuroleptics, as well as their side effects, has usually been understood in terms of their dopamine D
2 receptor activity. It is being increasingly realized that treatment may be optimized further by combining a high level of serotonin 5-HT
2 receptor blockade with low to modest levels of dopamine D
2 receptor blockade (
1–
3). The combination of 5-HT
2 with D
2 blockade provides antipsychotic treatment in which patients have lesser extrapyramidal side effects, greater improvement in negative symptoms, and perhaps even better improvement in positive symptoms in refractory cases (
3,
4). It has also been claimed that the superior efficacy of clozapine may actually result from its high level of D
4 blockade in combination with low to modest D
2 blockade (
5). Thus, current theories point toward a beneficial effect of high 5-HT
2 and high D
4 blockade in combination with low to modest D
2 blockade.
Loxapine has traditionally been considered a typical neuroleptic, but its pharmacological properties are rather atypical. Several in vitro studies (
1,
6,
7) have shown that its 5-HT
2 affinity is higher than its D
2 affinity. While the degree to which its 5-HT
2 affinity exceeds its D
2 occupancy depends on the particular technique and study (e.g., Meltzer et al. [1] reported a 5-HT
2/D
2 affinity ratio of 2.6; Leysen et al. [7], 6.8; and Singh et al. [6], 3.9), all reports agree that loxapine has a higher in vitro affinity for 5-HT
2 receptors than for D
2 receptors. Furthermore, in vivo animal studies show that loxapine, like clozapine but unlike other typical neuroleptics, down-regulates 5-HT
2 receptors in the prefrontal cortex, an outcome attributed to its prominent 5-HT
2 blockade (
8,
9). At the same time, loxapine is a very prominent D
4 antagonist, its D
4 affinity is higher than that of clozapine, and even its D
4/D
2 affinity ratio is comparable to that of clozapine (
6,
10).
However, there are significant differences between data from in vitro studies and the actual behavior of these drugs in humans. First, the affinity of a drug as determined in vitro varies with the assay and the conditions under which it is ascertained, and these may lead to widely varying results between laboratories (
10). Second, in vitro, one usually measures the efficacy of the parent drug itself. However, in vivo, the drug is metabolized, and the metabolite may have a pharmacological profile different from that of the parent compound and may reach a concentration higher than that of the parent drug. Third, two drugs with equal affinity in vitro may not penetrate the brain in the same fashion and therefore may give rise to different levels of occupancy in vivo. Finally, the net functional effect of a drug can be determined only in the context of clinical doses. A drug may have a higher relative affinity for 5-HT
2 than for D
2 receptors; however, if it is used at doses at which both of the systems are saturated, there may be no functional difference in the relative 5-HT
2 and D
2 blockades (
3).
Given evidence for the in vitro atypicality of loxapine, we were interested in documenting its in vivo receptor effects. We present here the results of the first systematic positron emission tomography (PET) study of the 5-HT2 and D2 occupancy profile of loxapine in humans.
METHOD
Ten patients (eight men and two women) aged 21–38 years participated in this study conducted in the Schizophrenia Division of The Clarke Institute of Psychiatry, Toronto. Patients were enrolled if they 1) had been taking a fixed dose of loxapine for 7 days or more; 2) had a DSM-III-R diagnosis of schizophrenia, delusional disorder (subject 2 in
table 1), or brief reactive psychosis (subject 5); 3) had not received a depot neuroleptic in the last 6 months; 4) were not taking any concurrent psychotropic medication except a benzodiazepine or an antiparkinsonian agent; and 5) had no concurrent substance abuse or dependence. Written consent was obtained from each subject on forms approved by the University of Toronto Human Subjects Review Committee. The main purpose of the study was to obtain the in vivo 5-HT
2/D
2 profile of loxapine in patients, for which this cross-sectional design is sufficient. However, because of the small size of the study group, the inadequate control over the treatment, and the fact that the patients were in various degrees of remission at the time of scanning, the design was limited in its ability to obtain reliable associations between receptor occupancy and clinical outcome. This is an important limitation of the study.
The 5-HT2 and D2 receptor status was assessed with the use of PET imaging with [18F]setoperone and [11C]raclopride, respectively, on a single day, after the patients had been taking a stable dose of loxapine for at least 7 days. The scans for dopamine D2 receptors were done 11–13 hours after the nightly dose of loxapine. The 5-HT2 receptor scans followed the D2 scans and were done 13–15 hours after the nightly dose. D2 receptor scans preceded 5-HT2 receptor measurements because the long half-life of [18F]setoperone would confound any subsequent [11C]raclopride studies. Both of the PET scans were obtained with a GEMS 2048-15B head-only scanner.
Seven of the patients (patients 1–7,
table 1) were part of an earlier brief report on the effects of loxapine on dopamine D
2 receptors (
11). The PET scans for D
2 receptors were obtained after the injection of 10 mCi (mean=10.02 mCi, SD=0.55) of high specific activity [
11C]raclopride (300–1600 Ci/mmol) according to a bolus-plus-infusion protocol described in detail elsewhere (
11,
12). Striatal and cerebellar regions of interest were drawn on two contiguous PET slices on a composite PET image with reference to a coregistered magnetic resonance imaging (MRI) scan (GE Signa 1.5-T scanner, T
2-weighted spin-echo sequence coregistered to the PET scan with the use of a surface-matching algorithm as implemented in ANALYZE 7.0 [Biomedical Imaging Resource, Rochester, Minn.]). To estimate the dopamine D
2 receptor binding potential (D
2BP) (which represents the total number of receptors available to the ligand, [
11C]raclopride, divided by the affinity of the ligand for the D
2 receptors [B
max/K
d]), we used a two-tissue compartment model that partitions the decay-corrected time activity counts obtained from the striatum into those which are specifically bound to the D
2 receptor and those which reflect the free radioligand and nonspecific binding (
13). The counts from the cerebellum were used as an estimate of the free and nonspecific binding in the striatum. The details of this method and its application to the determination of D
2 receptor occupancy have been described earlier (
12). This method yields a test-retest standard deviation of 6% and has been standardized to have an interrater and intrarater intraclass correlation coefficient, type III (ICC-III) greater than 0.95.
To estimate receptor occupancy we used an age-corrected baseline value derived from a pool of 12 neuroleptic-naive patients with DSM-III-R-defined schizophrenia. Loxapine-induced D
2 receptor occupancy was calculated as (D
2BP
Bas–D
2BP
Lox)/(D
2BP
Bas), where D
2BP
Bas is the age-corrected D
2BP baseline, and D
2BP
Lox is the D
2BP for the dopamine D
2 receptors in patients taking loxapine (
11). The absence of a subject's own baseline values introduces a potential error; the error is expected to vary from 0% to 9% for patients with 50% occupancy and from 0% to 4% for patients who have 80% occupancy (
11).
The 5-HT
2 scans were obtained by using a bolus injection of 5 mCi (mean=5.05 mCi, SD=0.22) of [
18F]setoperone according to the method developed and standardized by Blin et al. (
14,
15). The [
18F]setoperone was synthesized with a modification of methods described by Crouzel et al. (
16). The 5-HT
2 occupancy was determined in the prefrontal cortex. Since there was no significant difference across the two hemispheres, the data were pooled. The prefrontal region of interest was drawn on the [
18F]setoperone scan with reference to a coregistered MRI scan, as described above. Five contiguous PET slices that showed the prefrontal cortex were included in the region of interest. The cerebellar region of interest was drawn on two contiguous slices.
To obtain an index of the 5-HT
2 receptors, we chose the ratio of frontal to cerebellar activity over the 65- to 90-minute time period. The cerebellum is practically devoid of 5-HT
2 receptors (
17), and studies in baboons as well as humans report no displaceable [
18F]setoperone binding in this region (
14,
15,
18). It can be shown that at a time when the binding of the radioligand is at pseudo-equilibrium, the prefrontal/cerebellum ratio represents (1+k
3/k
4), or (1+B
max/K
d); k
3 and k
4 refer to rate constants that reflect ligand transfer between the free and specific compartments in a three-compartment model (
19). While this method does not permit an independent determination of B
max and K
d, it provides the binding potential for 5-HT
2 receptors (5-HT
2BP), an index that can be validly used for semiquantitative and within-study comparisons (
19). The details of this method have been described elsewhere (
20), and as operationalized in our laboratory, it yields an average test-retest deviation of 6%–7% and an acceptably high interrater reliability (ICC-III >0.95 for all regions) (
20).
Since these patients were already being treated, it was not possible to measure their baseline 5-HT
2BP. In the absence of this baseline, we used the age-corrected 5-HT
2BP obtained from 26 age-matched normal control subjects. Controlling for age effects is necessary, since age causes a definite decline in the number of 5-HT
2 receptors (
21), as was observed in this group of normal control subjects and has been observed in patients with schizophrenia (
22). The use of normal control subjects is justified by the fact that two studies that have systematically compared 5-HT
2 receptors in schizophrenic subjects versus normal subjects have reported no statistically significant difference (
22,
23). The loxapine-induced 5-HT
2 occupancy for a given subject was calculated as 100×(5-HT
2BP
Bas–5-HT
2BP
Lox)/(5-HT
2BP
Bas), where 5-HT
2BP
Bas is the age-corrected 5-HT
2BP for the drug-free state (obtained from the pool of normal subjects), and 5-HT
2BP
Lox is the measured 5-HT
2BP for patients taking loxapine.
The receptor occupancy data were related to plasma levels of loxapine and its metabolites 7-hydroxyloxapine and 8-hydroxyloxapine, which were ascertained at the time of each scan. The plasma levels were assessed by means of a previously standardized procedure in which high-performance liquid chromatography was used (
24). Plasma concentrations were estimated by comparing peak height ratios of each analyte with the internal standard and standard curves and with quality control samples analyzed during the same analytical run.
The relationship between an antagonist drug and the receptors that it occupies can be defined by the equation %ROcc=DConc/(ED50+DConc), where %ROcc is the percentage of the available receptors that are occupied by the drug, DConc is the concentration of the drug, and ED50 is a constant that equals the level of the drug required to occupy 50% of the available receptors. DConc should represent the concentration of the drug in the synapse. Since there is no easy way to measure the synaptic concentration of the drug in patients, one can use dose and plasma level as functional surrogates. However, it should be kept in mind that even plasma levels reflect synaptic concentrations only indirectly. Slight changes in the protein binding of an antipsychotic, without any change in total plasma concentration, may result in substantial differences in the levels of the freely available drug in the synapse. The observed receptor occupancy was related to the administered dose and measured plasma levels with use of the above equation implemented in SPSS for Windows (SPSS Inc., Chicago).
RESULTS
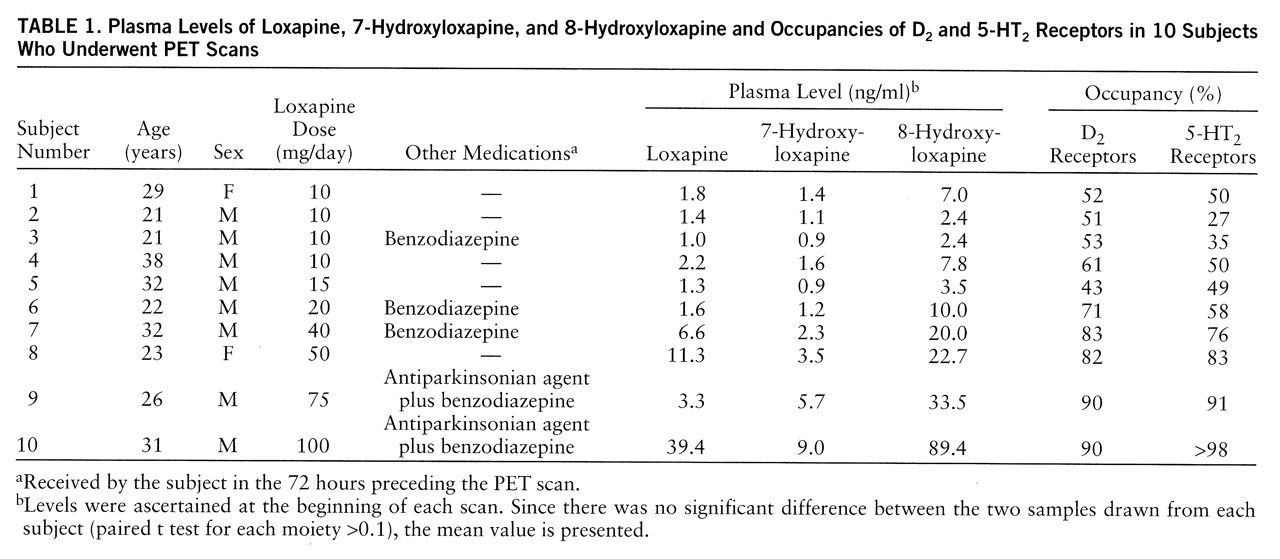
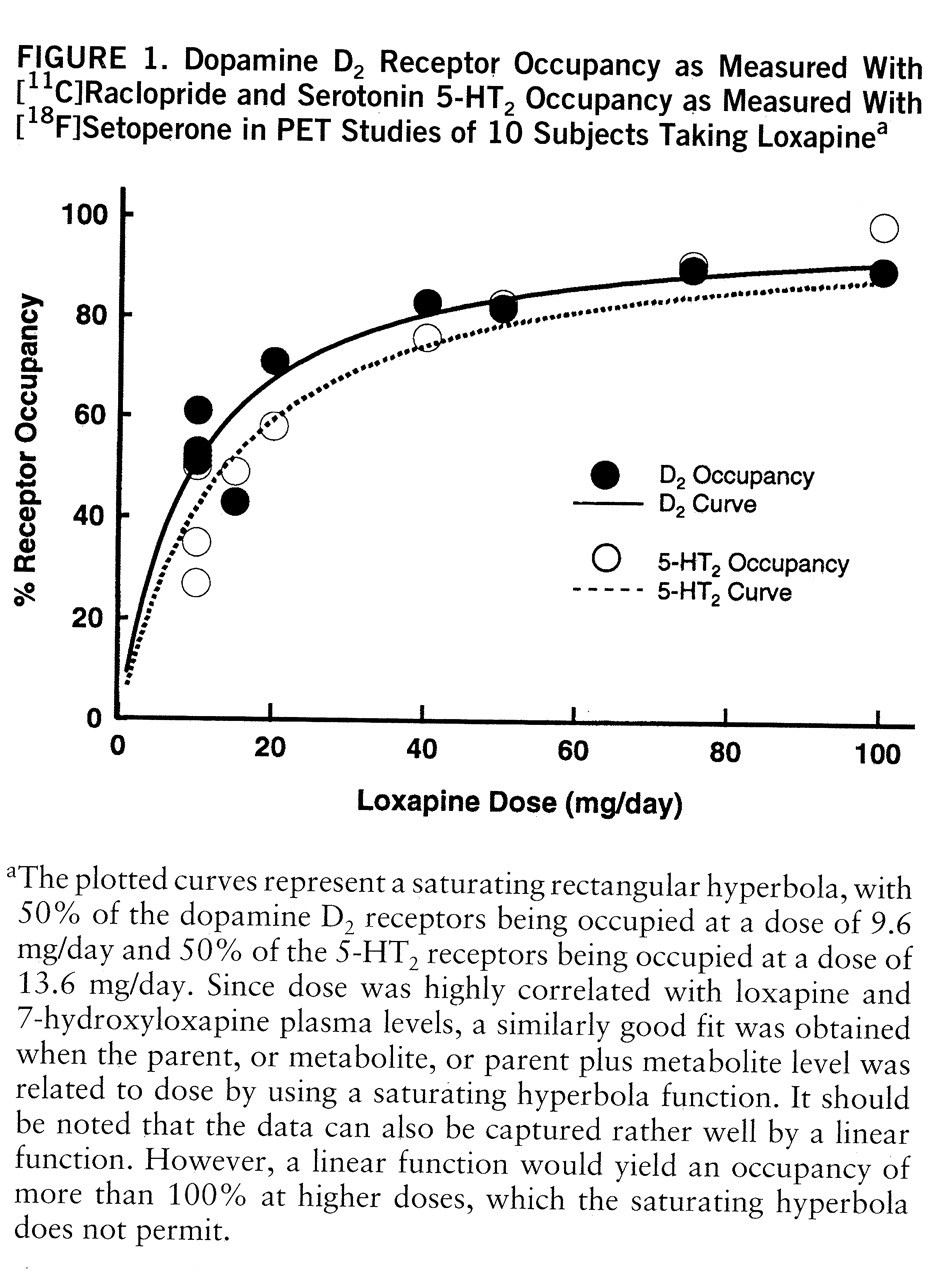
Data from all of the subjects studied are presented here. Dose significantly predicted loxapine and metabolite levels (loxapine levels in ng/ml=0.24×loxapine dose in mg/day [linear fit with no constant: F=23.7, df=1,9, p<0.001, R2=0.72]; 7-hydroxyloxapine levels in ng/ml=0.08×loxapine dose in mg/day [linear fit: F=370.6, df=1,9, p<0.001, R2=0.97]; 8-hydroxyloxapine levels in ng/ml=0.66×loxapine dose in mg/day [linear fit: F=79.9, df=1,9, p<0.001, R2=0.89]). The 7-hydroxyloxapine levels were, on average, 40% of the level of the parent compound, while the average of the 8-hydroxyloxapine levels was nearly 250% of that of the parent compound. The levels of the parent compound and the two metabolites were very highly correlated (for loxapine and 7-hydroxyloxapine, Pearson's r=0.87; for loxapine and 8-hydroxyloxapine, r=0.95; for 7-hydroxyloxapine and 8-hydroxyloxapine, r=0.97). The loxapine levels (not the metabolite levels) of one subject (subject 9) were clearly lower than expected and were reanalyzed to rule out any technical errors. He had, perhaps, an idiosyncratic metabolic profile, although the D2 and 5-HT2 occupancies were quite consistent with his dose.
The D
2 and 5-HT
2 occupancies are shown in
table 1. The relation between occupancy and dose is captured by a saturating hyperbola depicted in
figure 1. As shown in this plot, the dose to occupy 50% of D
2 receptors was 9.6 mg/day (95% confidence interval=7.2–12.0); the dose to occupy 50% of 5-HT
2 receptors was 13.6 mg/day (95% confidence interval=9.8–17.3). Loxapine is equipotent at the 5-HT
2 and D
2 receptors, since within a subject the D
2 and 5-HT
2 occupancies were not significantly different (t=1.72, df=1,9, p=0.12, paired t test), and the doses of loxapine to occupy 50% of D
2 and 50% of 5-HT
2 receptors were statistically indistinguishable.
DISCUSSION
Our study demonstrates that loxapine induces equivalent 5-HT2 occupancy and D2 occupancy as measured by [18F]setoperone and [11C]raclopride, respectively. We discuss these results in three conceptually different sections. First, we discuss the results of the in vivo 5-HT2 and D2 occupancies of loxapine in the context of the in vitro findings. Next, we compare the in vivo 5-HT2/D2 results of loxapine to those of the more widely accepted atypical neuroleptics clozapine and risperidone. Finally, we discuss these findings in light of our current knowledge regarding the 5-HT2/D2 receptor systems and their role in conferring atypicality upon antipsychotic action. We end the discussion with some suggestions for innovative pharmacological combinations that may be able to utilize the pharmacological profile of loxapine.
The first issue highlighted by our findings is the difference between the in vitro and in vivo results with loxapine. In vitro data from three different sources (
1,
6,
7) all show that loxapine has a two- to five-times higher affinity for 5-HT
2 receptors than for D
2 receptors. If the in vitro affinity applies in vivo, one would expect that the dose of loxapine that gives 50% D
2 occupancy would give anywhere between 66% and 80% 5-HT
2 occupancy. In our data we found no support for this. Loxapine's superior affinity for 5-HT
2 (versus D
2) in vitro was not observed in vivo.
Stockmeier et al. (
25) found a similar discrepancy between in vitro data and findings in rats. Loxapine showed a >2.5 times higher affinity for 5-HT
2 versus D
2 receptors in vitro (
1), but this was reduced to <1.5 times higher in vivo (
25). Stockmeier et al. speculated that loxapine's metabolites may have a higher affinity for the D
2 receptor, thus obscuring the in vitro superior affinity for 5-HT
2 of the parent compound. Our data support this contention of Stockmeier et al., since loxapine was extensively metabolized to hydroxy metabolites in the patients. In particular, the patients displayed high levels of 7-hydroxyloxapine, a metabolite that has a five times higher affinity for the D
2 receptor in comparison with the parent compound (
26). The other metabolite, 8-hydroxyloxapine, was found in higher quantities; however, it is relatively inert at the D
2 receptors (
26). Therefore, in all likelihood, the higher affinity of the 7-hydroxyloxapine metabolite for D
2 receptors may have diminished the in vitro superiority of the parent compound for 5-HT
2 (versus D
2).
The second issue of importance is the relationship of the loxapine findings to PET studies of risperidone and clozapine. Nordström et al. (
27) reported that patients taking clozapine exhibited 85%–94% 5-HT
2 occupancy even at low doses, while their D
2 occupancy varied from 20% to 67%. In a series of patients scanned with use of the methods described here, we observed that over the range of 2–12 mg/day, risperidone shows greater 5-HT
2 than D
2 occupancy. While at lower doses the difference between 5-HT
2 occupancy and D
2 occupancy may be as large as 15%–20%, at higher doses (>6 mg/day) the difference between occupancies is minimal, since both of the systems are near saturation. Therefore, the two atypical neuroleptics risperidone and clozapine show not only a high 5-HT
2/D
2 affinity ratio in vitro (
7,
28,
29) but also a high 5-HT
2 occupancy with a concomitant lower D
2 occupancy at clinical doses. This may help us understand why loxapine, despite having a high 5-HT
2/D
2 affinity ratio in vitro, has not been associated with atypical clinical benefits. While it does have a potential for producing high 5-HT
2 blockade, it does so only at doses that give a high degree of D
2 blockade.
If these suggestions regarding the reasons for atypical efficacy (that is, high 5-HT
2 occupancy with modest D
2 occupancy) are correct, then it would seem that augmenting the 5-HT
2 action of loxapine at a dose at which its D
2 occupancy is low should lead to the atypical benefits of atypical neuroleptics. Loxapine may be an opportune agent for augmentation, because it shares with clozapine a high affinity for the dopamine D
4 receptor (
6,
30) along with affinity for the 5-HT
3 receptor (
31) and 5-HT
6/5-HT
7 receptors (
32,
33). The exact contribution of these receptors to clozapine's uniqueness is not known (
6,
31–
33). However, given that loxapine also exhibits these properties, it is reasonable to hypothesize that augmenting the 5-HT
2 profile of loxapine with an add-on 5-HT
2 blocker, at a dose of the drug that provides modest D
2 blockade (10–25 mg/day), would give it a profile very similar to that of clozapine and other atypical neuroleptics.
The main hurdle in testing this hypothesis is that there are no specific 5-HT
2 antagonists available for regular human use. However, to provide a practical alternative, we have investigated cyproheptadine, an over-the-counter medication that is known to be a potent 5-HT
2 blocker in vitro. We found that 12–18 mg/day of cyproheptadine produced more than 85% 5-HT
2 blockade, as measured by the methods described above, in normal subjects (
34). A combination of 10–25 mg/day of loxapine and 12–18 mg/day of cyproheptadine should provide a clozapine-like profile not only at the D
2 and 5-HT
2 receptors but also at the D
4 and other serotonin, muscarinic, and histaminergic receptors. This combination needs to be tested in a clinical trial.
In summary, loxapine shows a higher affinity for 5-HT2 receptors than D2 receptors in vitro, but in humans the relative 5-HT2 superiority is lost. This may result from the potent action of its metabolite 7-hydroxyloxapine at D2 receptors, which may explain why loxapine, despite a very clozapine-like profile in the test tube, is not clinically an atypical neuroleptic. However, since loxapine shares several pharmacological properties with clozapine, it raises the possibility that if a low dose of loxapine (10–25 mg/day) is combined with a prominent 5-HT2 antagonist—thereby reinstating the 5-HT2 superiority that is lost in vivo—it may provide atypical antipsychotic benefits.