CRITIQUE OF NEURODEVELOPMENTAL HYPOTHESES OF SCHIZOPHRENIA
There are at least six broad major distinctions that are relevant to current theories of the causation of schizophrenia: 1) homogeneous or heterogeneous, 2) genetic or environmental, 3) single or multiple factors, 4) static or progressive, 5) short or long latency, and 6) developmental or nondevelopmental. Although these six dichotomies are independent of one another, so that all the combinations are possible, the analysis to follow is primarily concerned with only the last three. Because of this focus, the discussion could be read as implying that schizophrenia is assumed to be a single disorder with a single cause, but no such strong claim is intended, and the unifying hypothesis put forward, although intended to apply to a majority of schizophrenic patients, is consistent with causation by different genes in different subgroups, with multifactorial (either polygenic or genetic plus environmental) causation in individual patients, and even with the existence of a relatively small subgroup of patients whose illness has a primarily environmental etiology.
In the interest of clarity it should be noted that “developmental” is used in the literature with two meanings. Classically, it has been used in a temporal sense to indicate that the time during which a pathogenetic process actively damages the nervous system is limited to fetal or perinatal life, but for some time the same term has also been used to refer to the elaborate neurobiological mechanisms controlling the process of development. If the term is used only in a static temporal sense, then disorders of neural development are by definition early and nonprogressive, but if developmental refers to the neurobiological processes themselves, several of which normally extend into adult life, then developmental disorders caused by defects in one or another of these processes can continue to exert active pathological effects postnatally, i.e., to progress. It is crucial to note that the terms “static” and “progressive” are being used here in a narrow and mutually exclusive sense to contrast early termination to continued operation of a pathogenetic agent, and not in a broad sense that would encompass all of the continually unfolding secondary consequences of the pathology.
The major developmental processes of the human telencephalon following closure of the neural tube are neuronal proliferation, astrocytic proliferation, neuronal migration, neurite (i.e., axonal and dendritic) proliferation, neuronal apoptosis, axon myelination, and neurite pruning. All of these processes begin during gestation, but neurite proliferation, axonal myelination, and neurite pruning exert their major effects during postgestational development. It is interesting that physiological neuronal apoptosis may also be a normal part of human aging in certain restricted brain regions, most notably the substantia nigra, zona compacta (
43,
44).
The current positive evidence for a neurodevelopmental pathogenesis of schizophrenia (in the static temporal sense of the term “developmental”) has been set forth in a number of reviews (
6,
26,
45,
46). It includes neuropathological reports of cytoarchitectural abnormalities that are interpreted as being developmental in origin; absence of normal cerebral left-right temporal lobe asymmetries; results of prospective longitudinal studies comparing “high-risk” children, one or both of whose parents have schizophrenia or schizophrenia spectrum disease, to “low-risk” subjects; epidemiological studies relating higher prevalence of schizophrenia to winter season of birth, second-trimester maternal influenza, first-trimester malnutrition, and obstetric complications; and finally, data from retrospective studies that show subtle motor and facial expression abnormalities, mild learning difficulties, and impairment of social skills to be already present years before overt illness. This large body of evidence will not be reviewed in detail because, while it makes a strong case for placing the beginnings of pathogenesis in the pre- or perinatal period, it does not exclude subsequent postnatal progression.
Given the large range of possible neurodevelopmental derangements that might be implicated in disease pathogenesis, the main neurodevelopmental hypotheses for schizophrenia set forth in the last 10 years (
14,
26,
47,
48) are relatively restricted and share three assumptions: 1) the primary pathogenetic defect is an
early derangement of the orderly development of the central nervous system that occurs in the pre- or perinatal period; 2) the period of active operation of the causative agent is of short duration, meaning that it is essentially
static; and 3) the behavioral consequences of this static process remain relatively
latent until long after the primary pathogenetic process has run its course. The various neurodevelopmental models hypothesizing an early, static, and long-latency defect differ from one another primarily in the relative weight given to genetic versus environmental factors within individuals and in the degree of emphasis on hetero- or homogeneity of the disorder. (There is one neurodevelopmental model hypothesizing a
late, static,
short-latency defect [
49], but discussion of that model will be deferred to the next major section of this article.)
There are two major problems with the models involving an early, static defect. First, the excessive extracerebral CSF found in many imaging studies of schizophrenia is difficult for these hypotheses to explain. Second, schizophrenia is an illness that typically manifests itself in late adolescence or early adult life with major, progressive, and usually irreversible deterioration from previous levels of functioning (
1), and attempts to explain such a devastating disease by a subtle, localized, nonprogressive prenatal genetic or environmental insult to the brain run counter to a large body of data on functional recovery after early brain lesions (discussed later).
Implications of Imaging Data
The progression of quantitative brain imaging from CT scanning and the VBR to whole-brain volumetry with MRI has made reliable quantitative measurements of intracranial volume and its compartments, extracerebral CSF, ventricular CSF, gray matter, and white matter feasible. Since 1991 a number of MRI studies of chronically ill schizophrenic patients using quantitative measurements of gray and white matter and CSF, and using either ratios or covariance to control for intracranial volume, have shown a significant excessive overall loss of brain tissue (
50,
51) or gray matter (
52–
55) or an increase in extracerebral (sulcal) CSF space (
52,
54–
56) or a decrease of brain tissue in frontal regions (
50,
52) or in anterior cerebral cortex (
57) or orbitofrontal and mesiotemporal cortex (
58). Negative quantitative MRI results have been reported by one group for whole brain (
59) and for prefrontal regions (
60).
These findings of extracerebral CSF increase and brain volume decrease have important implications for any model of the neuropathogenesis of schizophrenia because of three fundamental principles of normal neural development: first, brain growth drives intracranial cavity growth; second, the brain grows outward from the ventricles; and third, intracranial cavity expansion is not reversible after skull sutures fuse. As a result, after brain growth reaches its maximum, intracranial size remains constant and any loss of brain volume leads to an equivalent increase in extracerebral and intraventricular CSF volume (see references
52 and
61 for more extensive discussions).
Given these principles, one can readily predict that diffuse loss of brain tissue limited to the pre- or perinatal time period will result in a smaller intracranial cavity and
persistent enlargement of the lateral ventricles but not to an increase in extracerebral CSF space. This is so because brain volume more than triples between birth and age 5 years, and any increase in extracerebral CSF caused by an earlier nonprogressive lesion would tend to be “filled up” by subsequent “outward” growth. Only a diffuse lesion resulting in a loss of brain tissue volume after maximum brain volume expansion has already taken place will result in an equivalent increase in total CSF space, both extracerebral and ventricular, and no change in intracranial volume; because the ventricles normally account for only about 10%–15% of total intracranial CSF volume (
50,
61), most of the compensating increase in intracranial CSF space will be extracerebral.
It is just because the ventricles can enlarge with both early and late tissue volume loss that cross-sectional studies, which look only at the ratio of lateral ventricular volume to total brain tissue volume (i.e., the VBR), can detect tissue loss but cannot distinguish between early and later time of occurrence. Only volume measurements that include extracerebral CSF volumes can determine whether or not there has been later volume loss.
Although the combination of excessive tissue volume loss and CSF space increase observed in patients with chronic schizophrenia might be a nonspecific consequence of psychosis, rather than reflecting an underlying causative neuropathological process, there are other imaging results that are not consistent with CSF volume expansion as an epiphenomenon of psychosis. The one published MRI study of new-onset schizophrenia in which tissue volume and both ventricular and extracerebral CSF were measured (
62) showed that, in relation to comparison subjects, the patients had a mean of 30 cc less brain tissue, 25 cc more extracerebral CSF, and 3 cc more ventricular CSF. Since intracranial volume did not differ between the groups, these results imply that the patients’ volume loss occurred in the time period after maximum brain volume expansion but before onset of overt illness.
Moreover, in a longitudinal “high risk” study that looked at the effects of both perinatal complications and genetic loading for schizophrenia on ratios of ventricular and sulcal (i.e., extracerebral) CSF volumes to intracranial volume, Cannon et al. (
63) found that while both of these ratios were positively correlated with genetic loading, ventricular enlargement was related to a history of perinatal injuries while sulcal enlargement was not. Since most of the subjects scanned in this large study did not have a diagnosis of schizophrenia themselves, and since the results held up when the overtly schizophrenic subjects were excluded, the positive relationship of extracerebral CSF volume to genetic risk for schizophrenia found in these subjects is not related to perinatal injury and cannot be a consequence of psychosis.
It should be emphasized that positive evidence for later brain volume loss in schizophrenia does not exclude early loss or failure of growth, even in the same patients. As already noted, early volume loss should be manifest as smaller intracranial volume, and a significant decrease in the latter was found in the frontal regions in a patient group who also demonstrated an overall excess of extracerebral CSF(
52,
61) and was also found in a meta-analysis of head size data from a large number of imaging studies (
64). Thus, although the rate of brain volume growth may be slower than normal beginning in early life (perhaps before birth) and also regress excessively in the period after onset of puberty, the critical point is that this later volume loss cannot be explained by the model of an early, static defect.
Problem of Latency of Clinical Illness Manifestation
The second major problem for neurodevelopmental models of schizophrenia that point to an early, static defect is the long latency between birth and the onset of clinically overt illness. To answer this objection, proponents of these hypotheses have sought parallels in established human and experimental animal models of developmental disorders. The most commonly proposed human models are developmental disorders such as dyslexia, in which the disorder is presumed to originate prenatally but not become manifest until the individual is faced with a later functional challenge (e.g., learning to read). In particular, the “lack of asymmetry” developmental model of schizophrenia (
4) draws heavily on the lack of cerebral asymmetry observed in both dyslexia (
65) and some but not all studies of schizophrenia (
66). One general problem with this analogy is that schizophrenia is an absolute deterioration from previous levels of functioning (
1), not a failure to keep pace with unaffected peers. There is, however, also a potentially important specific difference between the anatomy of schizophrenia and that of dyslexia; in schizophrenia the lack of asymmetry appears to result from a
reduction in volume of the side that is normally larger (see the raw data of Bilder et al. [
66]), while in dyslexia there is an
expansion of the side that is normally smaller (
65).
Other advocates of developmental models hypothesizing a genetic and/or environmental early, static defect (see, for instance, references (
14,
46, and
48) have looked to a combination of human neuroanatomical data (e.g., late myelination of parahippocampal regions[
67]) and animal models (
46,
68–
71) for plausible explanations or examples of late deterioration of function after static acquired pre- or perinatal lesions. The findings of Goldman (
72) in monkeys with prefrontal lesions acquired in infancy or the later juvenile period are frequently cited as illustrating just such a pattern, but a careful examination of those results does not support this interpretation. In that study, lesioned monkeys were repeatedly tested on a delayed alternation task at approximate ages 15 and 30 months (for lesions acquired in infancy) or at approximate ages 34 and 43 months (for lesions during the juvenile period). The performance of the early-lesioned animals was somewhat worse than that of control subjects at 15 months but did not change significantly from 15 to 30 months; what changed was the control subjects’ performance, which
improved. Thus, the gap between the performance of the lesion and control groups present at 15 months had widened by 30 months, owing to improvement by the control subjects rather than any overall deterioration in the lesioned animals (
72). It should be noted that the performance of the monkeys with early lesions was always superior to that of the monkeys with later lesions, implying some degree of compensatory reorganization (
73). These conclusions, which are the same as those drawn by the original author (
74), make it clear that this study does not provide an example of late functional deterioration after an early static lesion.
More generally, neurobiologists have had a long-standing interest in the consequences of early brain lesions, and the issue has been extensively studied. As originally noted by Kennard (
75), the developing central nervous system is able to compensate to a considerable degree for the deleterious effects of exogenous insults, even quite major ones, such as necrosis of a whole hemisphere (
76), so that the presumption is that if the fetus or infant survives and if the external agent stops being active (e.g., chronic seizure activity does not develop [[
77, p. 348]), then compensation will take place and, with certain exceptions, the earlier the insult, the greater the compensation (
78). The pattern of compensation is one of a relative sparing of early-maturing functions followed by a relative deficit of later-maturing functions (
79). There are experimental models in which early lesions are worse than later lesions (
80,
81), but in general these are not models of early lesions followed by a late deterioration of function from previously achieved levels.
One apparent exception to this conclusion that there is a dearth of late-deterioration models is a series of studies by Lipska and colleagues of pharmacologically induced neonatal ventral hippocampal lesions in rats (
68–
70). Such lesions are reported to lead to late-developing (postpubertal) vulnerability to both stress and medications (amphetamine, apomorphine, and others) that varies significantly in severity between one strain of rats and another (
69). This model does address the long-latency problem of the static-defect developmental hypothesis and also shows an intriguing interaction between an external injury and the genetic anlage. Nevertheless, careful analysis of the reported data suggests that in the experiments that were limited to environmental (i.e., nonpharmacological) stresses or stimuli, the model does not demonstrate spontaneous late deterioration of function after a static, early lesion.
On certain measures of behavior induced by environmental stresses, rats with ventral hippocampal lesions acquired on postnatal day 7 showed greater differences from control subjects, given sham operations, when tested on postnatal day 56 than they had when given the same tests on day 35 (
68). However, the data do not show that the absolute level of function of the lesioned rats got worse from day 35 to day 56. For example, in the case of what is known as prepulse inhibition, in which the startle response to a loud noise is attenuated by an immediately preceding small tone, on day 35 the unmedicated sham-operation rats had about 29% prepulse inhibition after a 4-dB warning tone, while the unmedicated lesioned rats had approximately an 18% prepulse inhibition, a nonsignificant difference (see figures 3 and 4 in reference
68). However, when the experiment was repeated at day 56, the sham-operation rats showed a 59% startle reduction and the lesioned rats a 30% reduction. This lesion-control difference was significant, but the changes over time came about not because the prepulse inhibition of the lesioned rats deteriorated from day 35 to 56, but because their performance improved only 12% while that of the control rats improved 30%.
A similar pattern is seen for another behavior, spontaneous locomotion. Unmedicated lesioned rats actually showed a slight
decrease in spontaneous locomotion between the ages of 35 and 56 days, but the decline was significantly less than that seen in control rats (see figure 3 of reference
70). Here too there was a relative but not absolute decline in function with age.
In another animal model paradigm of schizophrenia that involves an early pharmacological lesion, it has been proposed that early lesions lead to a neural disinhibition that results in delayed secondary excitotoxicity, which in turn leads to cortical volume loss (
71). This model can account for both the evidence that there are early brain lesions in schizophrenia and that there is later brain volume loss, but it deals with the lack of gliosis in the cortex of schizophrenic persons by simply concluding that this particular secondary excitotoxic process does not result in substantial persistent gliosis (
71).
Thus, if the issue is whether schizophrenia is neurodegenerative in the classic sense or neurodevelopmental in the static temporal sense, then it must be concluded that the combination of lack of gliosis with late loss of cortical volume poses serious problems for both hypotheses. If, however, the term “developmental disorder” is extended to encompass defects in one or another of the developmental processes that have prenatal origins but exert their major effects during childhood, adolescence, and even early adult years, then a hypothesis compatible with both the imaging and neuropathological lines of evidence is possible.
OUTLINES OF A UNIFYING HYPOTHESIS: SCHIZOPHRENIA AS A PROGRESSIVE DEVELOPMENTAL DISORDER
In light of the previous discussion, a unifying pathogenetic theory for schizophrenia that is consistent with all of the data requires a process that 1) begins prenatally; 2) progresses until it reaches a critical threshold, typically in the second or third decade; 3) causes progressive brain volume loss at a rate that is maximal in the first two decades and slows down with age; and 4) does not cause persistent gliosis (at least as detectable with current methods). Because the neuropathological data are contradictory as to whether there is actually cortical neuronal loss (
10,
12) or simply atrophy (
21,
82) and because atrophy (i.e., neuritic pruning) could be either pathological in itself or a manifestation of compensatory neural plasticity (
83), several variants of a model satisfying these criteria are presented.
One possible pathogenetic model is that of excessive neuronal apoptosis. Cell death, including neuronal death, can occur by two morphologically distinguishable mechanisms, necrosis and apoptosis (
84,
85). Necrosis in the central nervous system leads to inflammatory changes and gliosis, while apoptosis, which does not cause inflammation, occurs normally in early development and, at least in certain regions, with aging (
43,
86).
Apoptosis can also be pathological, arising either as a normal response to brain injury of many types or as the result of an intrinsic defect in its own regulation (
85). Many pathogenetic stimuli can trigger either necrosis or apoptosis, and the outcome seems to depend on the intensity of the stimulus (
87). Exogenous insults that cause apoptosis of neurons may or may not also cause proliferation (i.e., gliosis) and/or apoptosis of astrocytes (
88). Pathological neuronal apoptosis resulting from a genetically determined intrinsic regulatory defect has been convincingly demonstrated in the nematode
Caenorhabditis elegans (
89), and the excessive neuronal death in this model, which results from a defect in the
ced-9 gene, can be partially blocked by expression of the homologous human gene
bcl-2 (
90). Moreover, “gene knockout” mice homozygously deficient in
bcl-2 show a substantial excess of postnatal degeneration of peripheral sensory, sympathetic, and motor neurons (33% less of the last type than found in control subjects by day 44 [
91]), while knockout mice deficient in the pro-apoptotic gene
bax show a 51% excess of neurons in the facial nucleus (
92).
Thus, it seems reasonable to suppose that defects in
bcl-2, bax, other pro- and antiapoptotic genes in the
bcl-2 family and in other apoptotic regulatory genes may also occur in humans and result in excessive (or deficient) neuronal apoptosis. What is harder to know, however, is whether or not excessive neuronal loss by this mechanism would necessarily be accompanied by glial proliferation. If not, even widespread neuronal loss would be difficult to detect neuropathologically owing to the rapidity of the apoptotic process, which for a single cell takes about 1 hour from onset of observable morphological changes to removal of any detectable residuum (
93). Moreover, if postnatal pathological neuronal loss can result from nongliotic apoptosis rather than necrosis, the absence of gliosis would no longer limit the time of occurrence of that loss to early in gestation, and since apoptosis may be much more pronounced in one neuronal system than another (
85), the major brunt of the deficit could be restricted to a single anatomically distributed system.
Such nongliotic pathological apoptosis in schizophrenia has recently been suggested by Akbarian et al. (
94) as a possible cause of abnormalities in distribution and number of subcortical plate neurons (albeit not cortical neurons) in brains of schizophrenic persons. That study showed normal numbers of cortical neurons but too few subcortical plate neurons in the white matter just below the cortex and too many in the deeper white matter. Because the number of cortical neurons was normal, it was inferred that the problem causing the abnormalities in subcortical plate neurons was not one of cellular migration, as had been postulated in earlier papers (
95,
96), but rather aberrant apoptosis. Interestingly, although the published statistical analysis of the data was limited to showing that the subcortical plate neuronal distribution was abnormal, the accompanying numerical data and figure make it clear that there has also been an excessive overall loss of subplate neurons. Furthermore, there was no evidence of excessive gliosis in the affected areas, even though subplate neuronal apoptosis does not normally begin until late in the third trimester (
77, p. 70). This is an important negative finding because the fetal brain can react with gliosis as early as the 20th week of gestation (
25) and certainly throughout the third trimester (
26).
Aberrant apoptosis is not the only possible progressive neurodevelopmental pathogenetic mechanism for schizophrenia. Two groups (
21,
82) have found neuropathological evidence of increased neuronal density without cell loss (i.e., neuronal atrophy) in the cortex of schizophrenic persons, suggesting that the basic pathogenetic mechanism may be reflected not in neuronal loss but in greater than normal neurite pruning. (Interestingly, in the
bcl-2-deficient mice mentioned earlier, many of the neurons remaining in the facial nucleus were shrunken [
91].) Moreover, since pruning is related to both plasticity and learning (
83), it is quite possible that some or all of the excessive neuronal atrophy in schizophrenia might not be pathological in itself, but rather the result of attempted compensation for an ongoing pathological process, be it aberrant apoptosis or pruning. (The initial suggestion that pruning might be pathological in schizophrenia [
49] was nonspecific as to whether the problem was too much pruning, too little pruning, or abnormal pruning. However, observations of generalized cortical neuronal atrophy in schizophrenia [][
21,
82] seem to exclude too little pruning as the cause.)
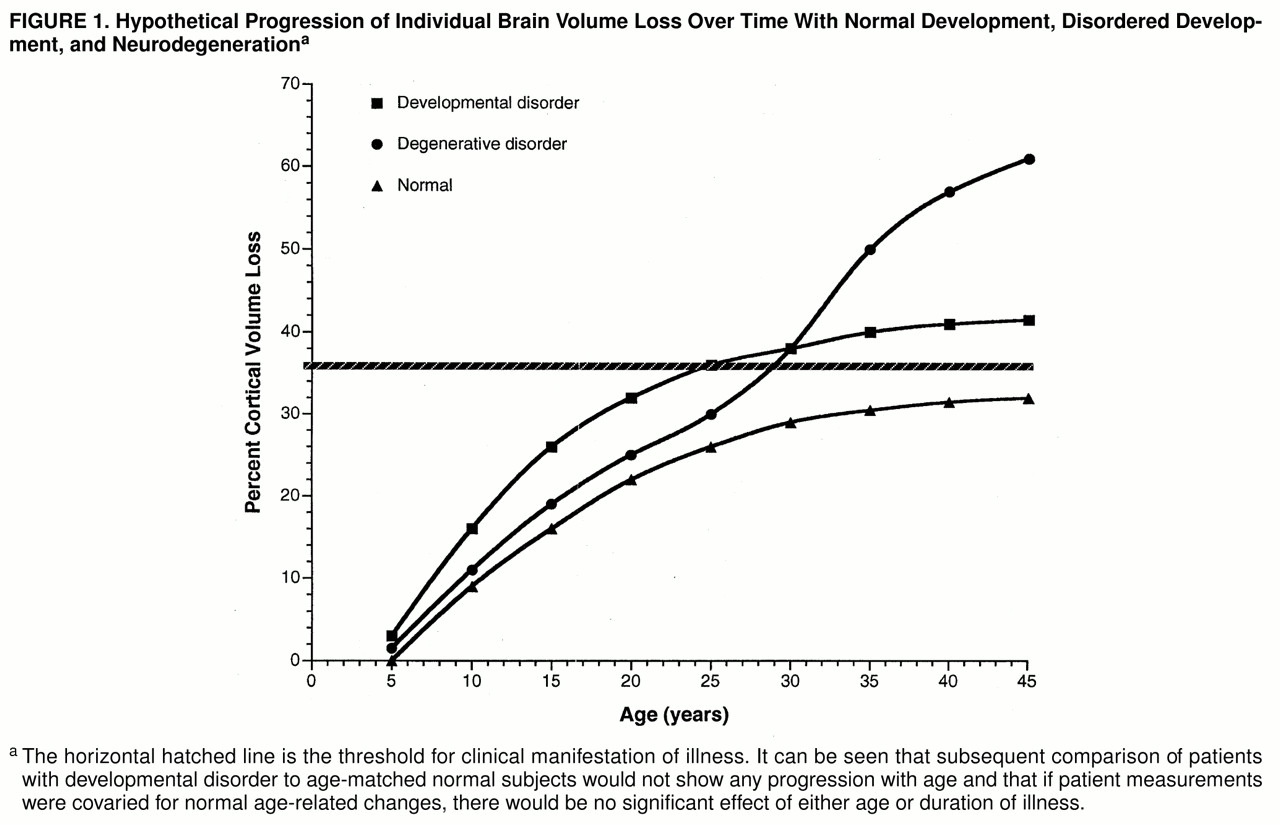
Since several MRI studies of brain changes normally occurring in childhood and adolescence show a rate of gray matter volume loss that is maximal beginning at about age 5 and then decreases asymptotically during adolescence (
97–
99), and since this volume change is believed to be due to neuronal pruning and not cell loss (
83), one can conclude that the rate of normal pruning gradually declines during childhood and adolescence. If pathological pruning were quantitatively greater but followed a similar asymptotic curve, the rate of loss might appear to plateau soon after crossing a threshold and producing overt illness. Figure 1 illustrates this by showing idealized patterns of cortical volume loss seen with normal age-related pruning, with hypothesized pathological pruning, and with a prototypical neuronal degenerative disorder in which the rate of neuronal loss is maximal at the time of onset of overt symptoms.
If the characterization of schizophrenia as a progressive neurodevelopmental disorder is correct, it is instructive to compare and contrast it to two other exemplars of this class of disorders, Rett’s syndrome and autism. Rett’s syndrome is a rare sex-linked disorder with marked female preponderance, in which neurological function is normal at birth but then deteriorates, initially rapidly and then more slowly until a plateau of rather severe disability is reached at about age 4 (
100). Most patients then survive well into adult life without any further worsening unless medical complications supervene. Brain and head size are normal at birth, but then brain growth lags severely behind what is expected, until by age 4 brain and head size stabilize at about 75% of normal size (
101). It is believed that most of the lag in brain growth during this period is due to a lack of neuritic proliferation (
100). Neuropathologically, there are no inflammatory changes and no gliosis (
102), but there are subtle changes indicative of prenatal onset. The disorder is thought to be of genetic origin, but it does not fit any classic pattern of inheritance (
100).
MRI volumetry in Rett’s syndrome shows a pattern of almost purely early volume loss. In a study of 20 Rett’s syndrome patients and 20 matched comparison subjects (mean ages, 9.7 and 9.0 years, respectively) the patients’ mean brain tissue volume was 297 cc
less than that of the comparison subjects, and the mean extracerebral and ventricular CSF volumes were, respectively, 8.5 cc and 3.2 cc greater than those of the comparison subjects (
101), meaning that 96% of the tissue loss is accounted for by a decrease in intracranial volume. This study was cross-sectional, and most of the patients were already beyond the active stage of the disorder, but serial measurements of head size make it clear that head size in Rett’s syndrome is normal at birth and the rate of growth begins to lag at about 2 months of age (
103).
Autism also is marked by apparent normality at birth and often up to 2 years of age, at which time there is an arrest or even regression of speech function and social interaction, but, unlike the case in Rett’s syndrome, no major impairment of motor function. The disorder gradually stabilizes over the next several years, although there may be an impressive development of certain cognitive skills. In direct contrast to Rett’s syndrome, there is in a number of cases actually excessive brain volume or weight (
104,
105). Neuropathological examinations of a limited number of brains from autistic patients show variable degrees of developmental abnormalities and localized increases in neuronal density but no generalized gliosis (
105,
106). Autism, like Rett’s syndrome, is thought to be genetic in origin, but it too does not fit any classic pattern of inheritance (
107).
MRI volumetry in autism shows a distinct pattern of early volume gain. In one study (
104), the patients’ mean brain tissue volume was 84.6 cc greater, and the extracerebral and ventricular CSF volumes were 6.8 cc and 7.7 cc greater, respectively, than those of comparison subjects, indicating that 85% of the gain in intracranial volume is accounted for by brain tissue growth. Here too the MRI study was cross-sectional, but serial head measurements again demonstrate normal head size at birth and subsequent excessive growth in a large subset (37%) of autistic individuals (
108).
The important parallels suggesting that Rett’s syndrome, autism, and schizophrenia are all progressive neurodevelopmental disorders are threefold: first, all are characterized by deterioration from previous levels of functioning; second, all show a leveling off rather than a lifelong progression of clinical deterioration; and finally, all share an absence of reactive gliosis on postmortem neuropathological examination. The important differences, suggesting that each is a distinct member of a common class of disorders, are seen not only in the characteristic clinical manifestations but in three very different patterns of abnormality on MRI volumetry: pure early loss in Rett’s syndrome, pure early gain in autism, and either primarily late (
62) or mixed early and late (
52,
61) loss in schizophrenia.