Hypothalamic-pituitary-adrenal (HPA) axis abnormalities have been described in a substantial proportion of patients with Alzheimer's disease (
1–
8). Interest in HPA axis function in Alzheimer's disease has been stimulated by evidence from animal studies that raised cortisol levels accelerate neuronal loss in the hippocampus (
9,
10). Furthermore, lesions of the hippocampus tend to increase HPA axis activity (
9). Hence, Sapolsky and McEwen (
9) proposed that hippocampal cell loss in Alzheimer's disease results in hypercortisolism, which in turn acts as a co-factor in further degeneration (the glucocorticoid cascade hypothesis).
If the glucocorticoid cascade hypothesis is relevant to the pathophysiology of Alzheimer's disease, then hypercortisolemia and nonsuppression of cortisol in the dexamethasone suppression test (DST) should occur in the same subjects, the rate of HPA axis dysfunction should be higher in more severely demented groups, and HPA axis dysfunction should worsen in individuals with disease progression. However, three studies have reported findings that are inconsistent with these hypotheses. First, Ferrier et al. (
11) reported that nonsuppression on the DST was not stable over time. Second, Weiner et al. (
12) failed to demonstrate any change in cortisol levels over a period of 1 year. Finally, Miller et al. (
13) reported a lack of association between nonsuppression and hypercortisolemia. Our aim in this study was to use the DST to examine longitudinal HPA axis function in Alzheimer's disease.
METHOD
Thirty patients who met the criteria for probable Alzheimer's disease of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (
14) were recruited for study. Twelve of these patients had very mild dementia as defined by a clinical dementia rating of 0.5 (
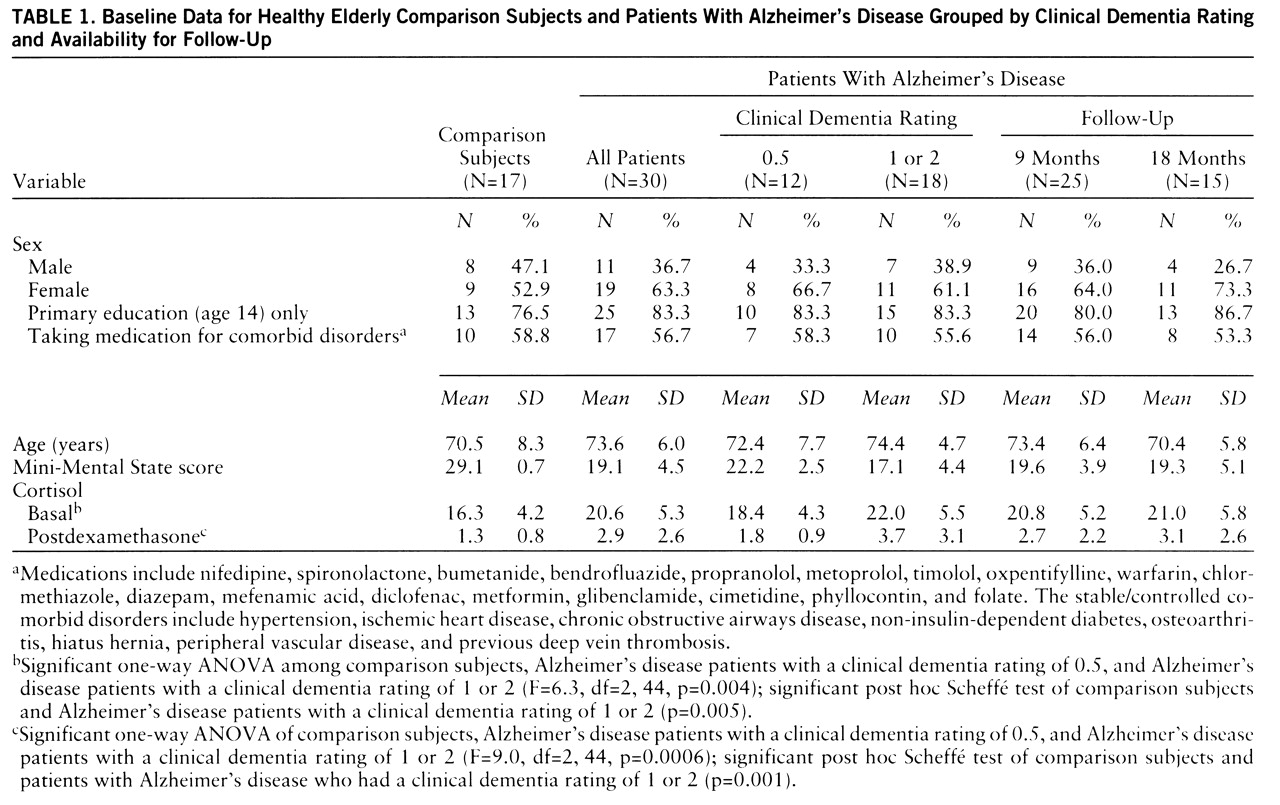
15); 11 had mild dementia (clinical dementia rating=1), and seven had moderate dementia (clinical dementia rating=2). All patients with clinical dementia ratings of 0.5 progressed to clinical dementia ratings of 1 after 12 months. A comparison group of 17 healthy elderly subjects was recruited from “active retirement” groups. Demographic characteristics of the two groups of subjects are shown in
table 1. Exclusion criteria were depression and substantial concurrent medical illness. The study was approved by the ethics committee of the Federated Dublin Voluntary Hospitals; written informed consent was obtained from each subject after he or she had been given a complete description of the study.
The DST was carried out as an outpatient procedure. Serum for basal cortisol estimation was taken at 8.30 a.m., and 1 mg dexamethasone was administered at 11:00 p.m. The next day, serum was taken at 8.30 a.m. and 4:00 p.m. Dexamethasone was always administered to patients with Alzheimer's disease by the people who usually took care of them, and postdexamethasone samples were not taken if there was any doubt about compliance. Cortisol was assayed by using a radioimmunoassay technique with inter- and intraassay coefficients of variation of 9% and 4%, respectively, and a sensitivity of 0.15–0.25 µg/dl. The DST was given again at 9-month intervals to the Alzheimer's disease patients. Nonsuppression was defined as any postdexamethasone cortisol level of ≥5 µg/dl (
16). (Postdexamethasone cortisol level refers to the higher of the two postdexamethasone levels obtained.) Hypercortisolemia was defined as any value greater than the mean for the comparison group plus two standard deviations.
The Mini-Mental State (
17) and the cognitive section of the Cambridge Examination for Mental Disorders of the Elderly (
18) were administered at the time of the initial DST and were repeated after 12 months.
Because postdexamethasone cortisol level has been shown to be log-normally distributed (
16), natural logarithmic transformation of the postdexamethasone cortisol levels was carried out. Analysis of variance (ANOVA) with post hoc Scheffé tests were used to compare group differences. Paired t tests were used to compare baseline and follow-up data. Pearson's product-moment correlation was used. Two-tailed levels of significance were applied throughout.
RESULTS
Cross-sectional findings. There were no nonsuppressors in the comparison group. There was no correlation between postdexamethasone cortisol levels and either age (r=0.16, df=16, p=0.55) or basal cortisol level (r=0.06, df=16, p=0.80). Four of the 30 patients with Alzheimer's disease were nonsuppressors. Six patients had basal hypercortisolemia (>24.7 µg/dl). Patients with Alzheimer's disease had significantly higher basal (unpaired t test, t=–2.8, df=45, p=0.007) and postdexamethasone (t=–3.5, df=45, p=0.001) cortisol levels. This reflected a difference between the patients with mild or moderate Alzheimer's disease and the comparison group (ANOVA,
table 1).
Postdexamethasone cortisol levels and Mini-Mental State scores were correlated (r=–0.44, df=29, p=0.02), as were both the basal and postdexamethasone cortisol levels and scores on the cognitive section of the Cambridge Examination for Mental Disorders of the Elderly (r=–0.42, df=29, p=0.02, for both cortisol measures).
At the initial assessment, only two of the patients with Alzheimer's disease who were nonsuppressors also had basal hypercortisolemia. Similarly, at the first follow-up assessment, only two of seven nonsuppressors also had basal hypercortisolemia. However, at both of these assessments, basal and postdexamethasone cortisol levels were correlated (r=0.58, df=29, p=0.001, and r=0.42, df=24, p=0.04, respectively).
Longitudinal findings. The mean drop in Mini-Mental State score over the two 9-month follow-up periods was 6.7 points (SD=5.4, range=1 to –21). Twenty-five patients had a repeat DST after 9 months and 15 after 18 months. At 9 months, seven of the 25 patients were nonsuppressors, compared with only four at baseline. However, there was no significant change in basal or postdexamethasone cortisol levels after either 9 months (t=1.8, df=23, p=0.09, and t=–1.4, df=23, p=0.16, respectively) or 18 months (t=–0.6, df=13, p=0.56, and t=–1.1, df=13, p=0.28, respectively). The majority of patients showed little or no change in cortisol levels. For those who did show a change, as many patients had a drop in cortisol levels as had an increase. There was no correlation between the change in Mini-Mental State score and the change in postdexamethasone cortisol level (r=0.1, df=24, p=0.76).
DISCUSSION AND CONCLUSIONS
This study addressed four questions: 1) Do hypercortisolemia and nonsuppression on the DST coexist within individuals? 2) Is the rate of HPA axis dysfunction higher in the more severely demented? 3) Does HPA axis dysfunction persist with follow-up? 4) In individuals, does dysfunction worsen with disease progression? Although the cross-sectional data supported positive answers to the first two questions, the longitudinal data are not consistent with positive answers to the second two questions.
When hypercortisolemia was defined according to Miller et al. (
13), the data supported a lack of association between nonsuppression and hypercortisolemia. However, the observed correlation between basal and postdexamethasone cortisol levels suggests that the apparent lack of association could be an artifact of putting a cutoff point on cortisol measures.
Using the clinical dementia rating and the cognitive section of the Cambridge Examination for Mental Disorders of the Elderly, we found a positive relationship between severity of dementia and cortisol levels. We studied subjects with a wide range of dementia severity (scores of 29 to 86 on the cognitive section of the Cambridge Examination for Mental Disorders of the Elderly), including a number of very mildly demented patients who required follow-up to confirm the diagnosis of Alzheimer's disease. None of the previous negative studies described such a group.
Consistent with findings of previous studies (
11,
12), we found no significant change in cortisol levels after 18 months and no consistent pattern of change within individuals.
The main limitation of this study was the lack of follow-up data on a number of subjects. It is worth noting, however, that all 12 patients with clinical dementia ratings of 0.5 had repeat DSTs. The follow-up period (9–18 months) was short. However, if the glucocorticoid hypothesis is relevant, then it should be possible to demonstrate consistent HPA axis dysfunction over short periods of time in the early stages of the disease process, when pathological changes preferentially affect the hippocampus (
19). Dexamethasone levels were not measured; therefore, reduced absorption or hypermetabolism in the patients with Alzheimer's disease cannot be excluded. Finally, it might be argued that complete compliance with the DST protocol cannot be ensured because it was performed as an outpatient procedure. Against this we argue that we avoided any potential effect that might occur due to hospital admission per se.
One explanation for our finding no association between cross-sectional and longitudinal measures is that the glucocorticoid cascade hypothesis is relevant only in the very early stages of the disease. Indeed, we have previously reported that the postdexamethasone cortisol level may be predictive of prognosis in very mild Alzheimer's disease but not in mild or moderate cases (
20).
Alternatively, the abnormal DST response may be related to such factors as the stresses of institutionalization, physical illness, and response to stress in the caregiver. Because some of these factors occur more frequently as the severity of dementia increases, they could explain the rate of HPA axis dysfunction in moderate and severe Alzheimer's disease.
In conclusion, we observed no association between the findings from longitudinal and cross-sectional measures of HPA axis dysfunction in Alzheimer's disease. The longitudinal data were inconsistent with a role for the glucocorticoid cascade hypothesis in the pathophysiology of mild and moderate Alzheimer's disease.