To be useful, a validating criterion must have both high sensitivity (to validate most syndromes that are true disorders) and high specificity (to invalidate most syndromes that are not true disorders). Only in this case can we be confident that syndromes meeting the validating criterion are likely to be true disorders.
Although the criterion of familial aggregation probably has high sensitivity (most true psychiatric disorders run in families), it has poor specificity because lots of things that run in families are not valid diagnostic entities. This point can be illustrated with the following scenario, in which physical rather than mental characteristics are central: A new disorder, “syndrome Z,” is proposed with three diagnostic criteria: 1) height over 6 ft, 2) red hair, and 3) a large nose. A family study of syndrome Z collects 100 affected individuals and 100 control individuals and then examines all first-degree relatives. A substantially higher prevalence of syndrome Z is found in the relatives of the affected individuals than in the relatives of the controls. On this basis, syndrome Z is declared to meet the “family study” validity criterion of Robins and Guze.
Since height, hair color, and nose size all “run in families,” a syndrome constituted of these three traits will, ipso facto, also be familial. The application of the “family study” criterion to syndrome Z will produce a false positive result. Because the preponderance of human psychological and physical traits are familial, such false positive results are likely to be common, undermining the value of the validating criterion of familial aggregation.
The Limits of Genetics as a Tool to Address Diagnostic Conundrums
Examining disorders one at a time in genetic designs provides nosologic information of limited value. This is not true when two or more disorders or criteria sets are examined. For example, in comparing two approaches toward diagnosing the same disorder, showing that one produces a higher degree of familial aggregation is useful information for the nosologist. Assume we have one well- established disorder–A–and a new disorder–B–that might be closely related. It would be of substantial nosologic interest to determine if, in a family study, disorder B occurs at elevated rates in the relatives of probands with disorder A. If the answer to this question is yes (as, for example, has been seen for schizophrenia and schizotypal personality disorder
[10] ), then there is evidence that these two disorders shared familial etiologic factors, which in turn might suggest a nosologic relationship. If the answer is no, diagnostic independence would be favored.
These same questions can also be asked, with greater conceptual clarity, by twin or adoption designs that can isolate genetic from familial-environmental effects. Twin studies—which assess all genetic effects together at the aggregate level—have in particular proven useful in determining the overall genetic relationship between different disorders, quantified in the statistic termed the genetic correlation. For example, the genetic correlation is very high for major depression and generalized anxiety disorder
(11) but lower for major depression and animal phobia
(12) and alcohol dependence and pathological gambling
(13) .
Although such results can
inform nosologists’ decisions, they cannot, in and of themselves, answer the fundamental nosologic questions. This is true for at least two reasons. First, taking twin studies as an example, these studies alone cannot address the issue how high a genetic correlation has to be to consider two syndromes to be subtypes of a single disorder or how low a genetic correlation has to be to consider the two syndromes to be independent diagnostic entities. Second, such studies alone cannot answer questions such as What should nosologists do if two disorders have closely related genetic risk factors but distinct environmental risk factors or are genetically distinct but respond to the same kinds of treatment? and Should genetic risk factors be given highest priority in such nosologic decisions? Such questions cannot be addressed by purely empirical means but require judgments about the relative importance, in a particular nosologic decision, of different potential validators
(14) .
However, family, twin, and adoption studies are now not the only approach in psychiatric genetics that can be applied to diagnostic conundrums. Linkage and association studies can provide information, respectively, about whether genomic regions or specific genes influence risk for more than one disorder. Will these newer methods prove of greater value to the psychiatric nosologist? Might it be possible that just as molecular genetics has been used in biology to help define species, it might be similarly used to define psychiatric disorders?
As an example of this approach, Berrettini
(15) reviewed evidence from linkage studies suggesting regions with linkage to both schizophrenia and bipolar illness. He wrote:
Review of these data indicates that there are five genomic regions that may represent shared genetic susceptibility of BPD [bipolar disorder] and SZ [schizophrenia]. As the genes underlying these confirmed linkages are identified, the current nosology must be changed to reflect the new knowledge concerning the shared etiologies of BPD and SZ.
There are at least two potential caveats to these claims: 1) different genes under the same linkage peak could influence liability to schizophrenia and bipolar illness, and 2) these regions of “joint” linkage might arise by chance, given the large number of chromosomal regions putatively linked to each disorder. However, some evidence, which is still quite preliminary, has suggested that individual candidate genes may be associated with both schizophrenia and bipolar illness (e.g., see references 16–18). In one such study, using a modest sample size, Hodgkinson et al.
(16) found several alleles at single-nucleotide polymorphisms in the
DISC1 gene that were significantly associated with schizophrenia (with odds ratios of 1.2 to 1.3) and other alleles in the same gene that were significantly associated with bipolar illness (with odds ratios varying from 1.1 to 1.2). What impact should it have on our nosology if one or more genes are shown definitively to be associated with both schizophrenia and bipolar illness?
The liability to multifactorial disorders such as schizophrenia and bipolar illness are almost certainly influenced by a large number of genes
(19,
20) . For the sake of argument, assume that each of these two disorders is influenced by variants at 20 different genes. What would it mean if verified findings emerged that one, two, or even five susceptibility genes were shared between these two disorders? At what point should we alter our nosology on the basis of such findings?
General medicine contains many examples of distinct diagnostic categories that share genetic risk factors. For example, genes that predispose an individual to essential hypertension
(21) will increase the liability to hemorrhagic stroke, myocardial infarction, and hypertensive cardiomyopathy. Mutations in the oncogene
BRCA1 increase risk for cancer of the breast, cervix, uterus, pancreas, fallopian tube, stomach, colon, and prostate
(22) . Yet, this evidence for “common genes” has not been used to support changes in the classification of these disorders.
Finding a small number of genes that influence susceptibility to two multifactorial disorders is not likely to provide definitive information for nosologists. Indeed, from a nosologic perspective, this information differs little from finding a modest genetic correlation between two disorders in twin studies.
A different situation would emerge if, as we identify susceptibility genes, we found that all or nearly all the genes that predisposed to disorder A also predisposed to disorder B. Such a finding, which would represent a confirmation at a biological level of results from family studies (that is, high levels of coaggregation) or twin studies (a high genetic correlation), would provide further evidence that the two disorders were closely related.
To illustrate a likely pattern of results that will emerge for pairs of related psychiatric disorders, it is informative to review what has been learned about the genetic relationship between the two major forms of inflammatory bowel disease—Crohn’s disease and ulcerative colitis. Most, but not all, family studies have indicated modest levels of coaggregation, with better evidence that rates of ulcerative colitis are increased in relatives of Crohn’s disease probands than vice versa
(23,
24) . Twin studies have suggested that both disorders are heritable. Monozygotic twin pairs where one twin has Crohn’s disease and the other ulcerative colitis are, however, rare
(23) . In a recent meta-analysis of 10 linkage studies of inflammatory bowel disease, suggestive evidence for linkage was found in six regions for Crohn’s disease and in only one region for ulcerative colitis
(25) . However, the single region of tentative linkage for ulcerative colitis (2q) was one of the six found for Crohn’s disease. Association studies have shown that variants in the best replicated susceptibility gene for Crohn’s disease (
CARD15 ) do not influence risk for ulcerative colitis
(24) . A high-risk haplotype identified in chromosome 5q31–33 predisposed to risk for Crohn’s disease but not ulcerative colitis. However, variants in the human leukocyte antigen region have been found that increase risk for both disorders
(24) .
What should a nosologist conclude from these results about the relationship between Crohn’s disease and ulcerative colitis? Some genetic risk factors are shared, and others appear distinct. Would this pattern of results—which may be common for moderately related complex syndromes in biomedicine—lead easily to a clear decision about the nosologic relationship between the two disorders? It seems unlikely that molecular genetics will bring the same clarity to the classification of complex diseases in medicine as it has for species in biology.
It is worth asking at this point whether we are at risk for adopting too “gene-centered” a view of psychiatry–of making too much nosologically of the modest effect sizes we are finding for individual genes. For example, severe sexual abuse in an epidemiologic sample of women increased the risk both for major depression and for drug abuse with odds ratios of 3.14 and 5.70, respectively
(26) . Although these figures are much greater than that seen in the studies showing association of the same gene with schizophrenia and bipolar illness, such findings have not lead to suggestions to modify our nosology to reflect shared etiologies of depression and substance abuse.
The Limits of Mendelian/Essentialist Models for Psychiatric Disorders
Advocacy for Essentialist Gene Models for Psychiatry
In a lecture on psychiatric genetics given by a leading academic psychiatrist in 1987, the speaker began by outlining the recently successful efforts at identifying the genes underlying several Mendelian disorders and then went on to say:
With the new advances in molecular genetics and linkage analysis, by the time we get to DSM-V, we will be diagnosing chromosome 4 schizophrenia, chromosome 14 schizophrenia, and chromosome 22 schizophrenia. [His choice of particular chromosomes was illustrative rather than data-based].
In expressing such optimism about the impact of advances in psychiatric genetics on our diagnostic system, this individual was also suggesting something more fundamental. In predicting that our diagnostic manual would contain subtypes of schizophrenia identified by chromosomal location, he was advocating an essentialist model in which abnormal genes would be the
defining feature of psychiatric disorders . Such an essentialist perspective assumes that psychiatric conditions are not man-made constructions but rather represent real disorders that exist out there in the world and can be defined by their underlying nature (or
essence )
(27) .
The essentialist model of disease assumes that diseases can be classified in the same manner that atomic elements can be classified. Gold, silver, and lead are all true and independent entities, each with a unique essence—in this case defined by the number of protons in the nucleus.
Mendelian diseases are defined by the genes that cause them. Just as discrete microorganisms cause particular infectious diseases, so discrete genes cause particular genetic diseases. Just as individual types of bacteria or viruses are true entities in the world, so genes could be seen as classic essentialist categories, each with its own essence, roughly analogous to the atomic elements.
Essentialist disease models are very attractive. They are conceptually simple, appealing, and easy to teach. They fit well into the traditional medical model, thereby supporting the status of psychiatry as a medical discipline. Further, if they are linked to biological causes, they provide support for an organic disease model where psychiatric disorders are understood as resulting from pathological processes in the brain. They appear legitimate to third-party payers who can accept them as “real” diseases.
Indeed, the claim that the two great historical success stories in our field—general paresis of the insane and pellagra—are essentialist diseases rests on their both resulting from highly discrete causes: Treponema pallidum and vitamin B 3 deficiency, respectively. In the adjoining discipline of neuropsychiatry, in the last 15 years, the essentialist disease status of a series of disorders has been legitimized by the localization and subsequent identification of the genes that caused them, including Huntington’s chorea, early-onset Alzheimer’s disease, and Wilson’s disease. No wonder some in psychiatry were suffering from “gene envy,” which expressed itself in their advocating what could be called an essentialist gene model (EGM) for psychiatric disorders.
Early Evidence on the Plausibility of Essentialist Gene Models for Psychiatry
Although appealing, is the EGM, as exemplified by Mendelian diseases, applicable to psychiatric disorders? Back in 1987, things already did not look good for this model, which depends critically on the etiologic link between gene and illness being so strong that the essence of the disorder is explicable by a disruption of gene function
(28) . Nothing in the twin, family, and adoption studies of psychiatric disorders suggested that they were nearly as genetically simple as Mendelian disorders. Despite much searching, no well-validated pedigrees had been found where a major psychiatric disorder is transmitted through generations as a classical Mendelian trait. The risk of illness in relatives of ill patients never resembled those expected from the simple laws of Mendel. Since 1987, further information on four fronts has all suggested the inapplicability of the EGM for psychiatric disorders. First, several high-profile linkage results in bipolar illness and schizophrenia
(29 –
31) that were in part predicated on Mendelian models of illness could not be replicated. Second, many linkage studies were performed for schizophrenia and bipolar illness, and no replicated evidence was found for genes with a Mendelian-like effect. Instead, it appears that these disorders are the result of at least a moderate number of genes that individually have small to modest effects on disease liability
(19,
20) . Third, supportive of the preliminary findings in psychiatry, work in model organisms demonstrated that genetic influences on the vast majority of behavioral traits were the result of many genes, each of a modest effect size
(32,
33) . Fourth, animal studies consistently showed that, in model organisms, genes that influence behavior typically impact on multiple phenotypes
(32) . This phenomenon, termed pleiotropy, is inconsistent with one central feature of the EGM—the existence of a one-to-one relationship between gene and disorder (like the one-to-one relationship in the periodic table between the number of protons in the nucleus and other characteristics of the element). Pleiotropy may be so widespread because it reflects the fundamentally opportunistic nature of evolution
(34) .
The Limits of Gene Discovery for Psychiatric Nosology, or “Once We Find the Genes…”
At a conference in 2004, the following conversation between two psychiatric genetics researchers was overheard:
By 2004, it had become clear to everyone in the field that no “Mendelian-like” genes for psychiatric disorders were likely to be found. Nonetheless, there is continued hope that advances in psychiatric genetics and particularly the identification of individual susceptibility genes will alter, in fundamental ways, our approach to psychiatric diagnosis. If we are able to find a “gene for” a particular psychiatric disorder, then we can work our way back up and—as predicted by the EGM—ground our diagnostic category on the firm foundation of a gene.
Categorical Gene Models and the Problem of Small Effect Size
One of the hopes expressed by these researchers is that new discoveries in psychiatric genetics will permit us to define the boundaries of psychiatric syndromes. They have expressed a second implicit expectation for the nosologic impact of psychiatric genetics research—that it will support categorical definitions of illness. A categorical view of psychiatric illness—that these disorders are discrete entities with distinct boundaries—can be contrasted with the perspective that these disorders are pathological ends of functional continua.
Advocates of a categorical perspective on psychiatric disorders suggest that they are discrete entities that are similar to biological species, such as whales and cows, or to man-made objects such as shirts and pants. Such entities have distinct boundaries. It is clear what is inside and what is outside. With categorical entities, the task of the nosologist becomes finding these boundaries or, as is oft said, “carving nature at its joints.”
Classical Mendelian disorders appear to be such categories. Clinically, these diseases appear to be discrete. In families with multiple affected individuals, there are typically no “spectrum” cases; individuals are either affected or unaffected. Perhaps genetic discoveries would uncover such clear forms of psychiatric illness.
Categorical models of disease, by definition, require discrete boundaries or what has been termed “points of rarity.” For genes to be useful in defining categorical disease entities, the etiologic effect of the gene must be large enough that it produces such a “point of rarity” between those who possess and those who lack the disease gene
(35) . Classically, this would be represented as bimodality in a distribution of liability—the joint that is to be carved. To successfully ground a categorical diagnostic system in pathogenic genes, the genes need to affect liability strongly enough that their impact is detectable above the background effect of other risk factors.
As reviewed earlier, prior evidence from linkage studies of psychiatric disorders and animal behavior genetics do not provide encouraging news for the EGM. The effect sizes of genes found in these studies have typically been small. In the last decade, individual susceptibility genes have been tentatively identified for psychiatric disorders. Therefore, we can examine the magnitude of their effect more directly. A review of positive meta-analyses of functional candidate genes for psychiatric disorders found odds ratios ranging from 1.07 to 1.57, with a median of ~1.30
(36) . (An odds ratio is the risk for a disorder given the presence of a risk factor—here a particular gene—divided by the risk for that disorder in the absence of exposure to the risk factor.) In schizophrenia, replicated evidence is now emerging for several genes that have been localized under linkage peaks
(37), in particular dysbindin 1 and neuregulin 1. A recent review of dysbindin studies suggested that the odds ratio of variants in the gene and risk for schizophrenia average around 1.50
(38) . For neuregulin 1, two recent replications reported odds ratios of 1.25 and 1.80
(37) .
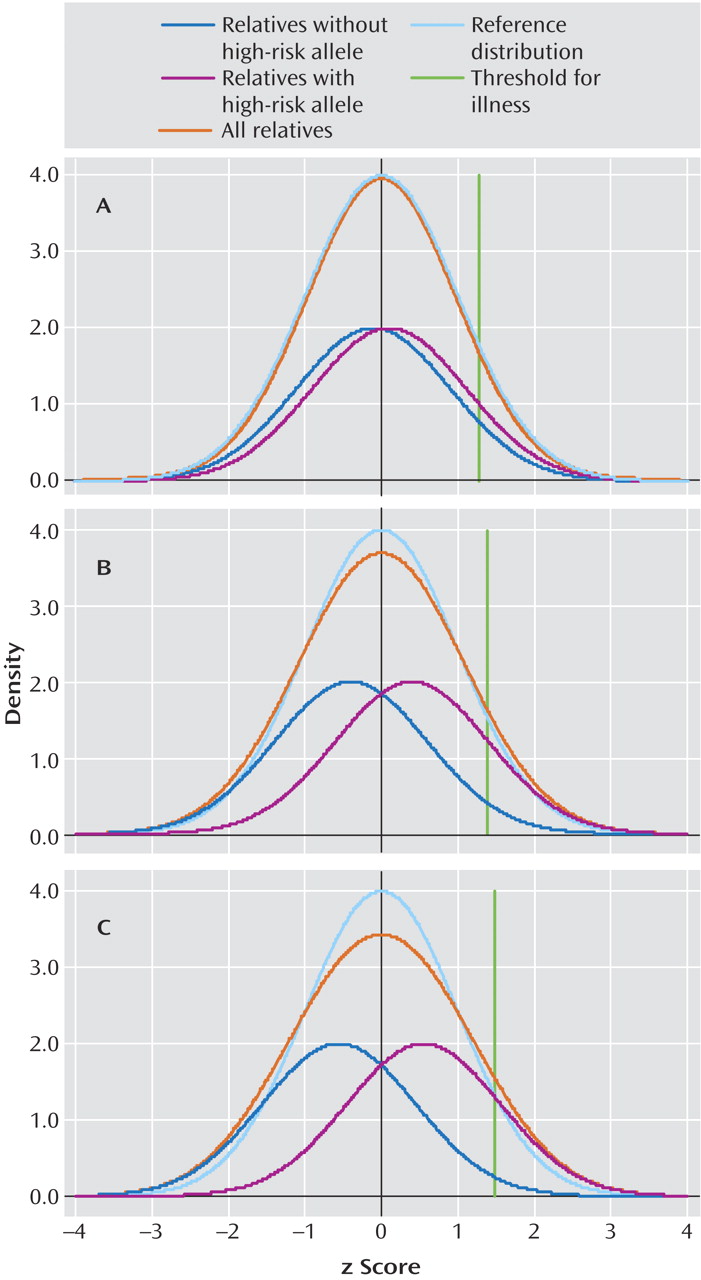
Figure 1 displays the liability distributions in a putative sample of first-degree relatives of individuals with schizophrenia, one-half of whom possess a high-risk copy (more technically allele) of a gene for schizophrenia. (The term “liability” here reflects the individual’s level of risk for the illness, with risk increasing as you move from left to right in each panel in
Figure 1 .)
Using plausible parameters (see
Figure 1 legend for further details), we have varied the magnitude of risk conveyed by the gene to produce odds ratios for the relationship between the high-risk allele and schizophrenia of 1.5, 5, and 10 in
Figure 1 panels A, B, and C, respectively. Each panel presents four different distributions of liability. The dark blue line reflects the liability distribution of relatives without the high-risk allele. The purple line reflects the liability distribution of relatives with the high-risk allele. The turquoise line reflects the “reference” liability distribution that would be seen if there were no individual genes of detectable effect and only background genetic and environmental variation that would be predicted to take the shape of a normal distribution. The orange line, which is the most important one, reflects the liability distribution of the population of all relatives and is simply the sum of the blue and purple line. In addition, the green line represents the cutoff point for illness. Individuals with liability above that threshold will develop illness.
The thought experiment we are here conducting is as follows: If we could measure liability directly in these relatives (although we would not know their individual genotype), could we cut cleanly (at nature’s joint) between those at high risk (depicted by the purple line) and those at low risk (depicted by the blue line)? That is, in the total population distribution (depicted by the orange line), do we see a clear point of rarity separating the two groups? For a gene with an odds ratio of 1.5 (
Figure 1, panel A), there is virtually no deviation in the population distribution from that predicted by a single normal distribution. Even with odds ratios of 5 or 10 (
Figure 1, panels B and C, respectively), all that is observed is a slight flattening of the distribution with no evidence for a point of rarity at which to divide those carrying and not carrying the high-risk allele.
Effect sizes are the range of those seen with genes that impact on risk for psychiatric disorder are too small to produce, on their own, syndromes with discrete boundaries.
Limits in the Concept of “The Gene” as a Tool in Psychiatric Nosology
Cracks in the Essentialist Image of Mendelian Disorders
While psychiatric genetics has sought to understand how genes contribute to psychiatric illness, cracks have appeared in the essentialist image of Mendelian genetic disorders. Examples have emerged that challenge a central feature of the gene-based essentialist disease model: the one-to-one relationship between gene and disease. Numerous instances have been found of mutations in different genes that all cause the same disease or similar diseases. Researchers have identified more than 60 chromosomal regions that lead to Mendelian forms of nonsyndromic hereditary deafness (where deafness is the only symptom)
(39) . At least 14 genes that produce chronic progressive hereditary ataxia have been identified
(40) . Three distinct genes produce Mendelian forms of early-onset Alzheimer’s disease
(41) . Many Mendelian human diseases arise from dysfunction in complex biological pathways that involve products from numerous distinct genes. Abnormalities in different genes in these pathways are likely to produce similar diseases.
Furthermore, mutations in the same disease gene can produce distinct phenotypes. Cystic fibrosis results from dysfunction in the
cystic fibrosis transmembrane conductance regulator . However, in addition to leading to classic cystic fibrosis, mutations in the this gene can solely cause congenital bilateral absence of the vas deferens, isolated idiopathic chronic pancreatitis, mild late-onset pulmonary disease, sinusitis, allergic bronchopulmonary aspergillosis, and possibly asthma
(42) . Dysbindin has been associated with schizophrenia in more than 10 independent samples
(38,
43) . However, mutations in this gene also cause Hermansky-Pudlak syndrome type 7, which is characterized by symptoms—oculocutaneous albinism, prolonged bleeding, and pulmonary fibrosis—that are apparently unrelated to those of schizophrenia
(44) . Mutations in
fibroblast growth factor receptor 3 produce a range of skeletal dysplasias with quite distinct phenotypes
(45) .
Even within medical genetics, essential disease models based on “the pathogenic gene” have become more difficult to sustain as evidence has mounted that different genes can cause the same disorder and abnormalities in one gene can cause different disorders.
Cracks in the Essentialist Image of the Gene
Few concepts in biology have generated as much controversy as the nature of “the gene”
(46 –
48) . Debates have focused on the validity of several different conceptualizations, including 1) a statistical definition as seen in population genetics or genetic epidemiology, 2) a latent “unit” controlling phenotypic inheritance as conceptualized by Mendel and Morgan, 3) the template for production of a unique protein, and 4) a discrete physical entity that is a specific piece of DNA with a particular chromosomal location. When psychiatrists think about grounding their essentialist diagnostic concepts on the firm foundation of genes, they focus on the third and fourth definitions—a specific “hunk” of DNA with a discrete biological function. We see the gene as a clear “natural kind”—a material entity that exists as a real, discrete unit in the world. In basing our “messy” diagnostic concepts on this natural kind—the gene—we hope that nosologic clarity will follow.
However, advances in molecular biology have undermined these simple definitions of the gene. The “one gene=one enzyme” hypothesis has been falsified. In the human genome, 75% of multiexon human genes are alternatively spliced with approximately 3.5 alternative forms of each gene
(49) . Neuregulin 1, one of the best supported susceptibility genes for schizophrenia, produces at least 15 distinct protein products
(50) . That is, with alternative splicing, the same gene, defined at the level of nucleotide sequence, produces different mRNA transcripts, which are in turn transcribed into different proteins. (So each transcript produces a unique protein, but one gene produces multiple transcripts.) If a multiply spliced gene contains a variant sequence in one of its alternately spliced exons, that variant will be present in some but not other proteins produced from the gene.
Many of these alternatively spliced genes have multiple promoters, with the result that different protein variants of a single gene are expressed at distinct times in different tissues. For example, the gene α-tropomyosin in the rat produces seven distinct proteins, two of which are expressed in striated muscle and one each in smooth muscle, myoblasts, fibroblasts, brain, and hepatomas
(48) .
The functional boundaries of the “gene” concept have been blurred by a phenomenon termed “gene sharing” whereby the same gene product serves dramatically different biological functions. For example, Piatigorsky
(51) has documented instances in which several metabolic enzymes have been “recruited” to also function as crystallins in the vertebrate lens.
A further uncertainty in the function of “the gene” arises from RNA editing—the posttranscriptional alteration of RNA sequence from that encoded in DNA
(52) . In some cases, such editing alters the structure of the expressed protein.
The physical boundaries of a “gene” are also becoming blurred. Key to the functioning of classic protein-transcribing genes is a series of control regions that influence the rate of transcription. Although such regions—termed promoters—exist immediately upstream of the coding region, researchers have found other control regions (enhancers and repressors) that are up to a million base pairs upstream or downstream and sometimes even in the introns of neighboring genes of unrelated function
(53) .
New variants of noncoding (nc) RNA have been discovered that further obscure the boundaries of what is meant by a “gene”
(54) . These ncRNAs can be classified into two broad groups: housekeeping ncRNAs and regulatory ncRNAs. Housekeeping ncRNAs are involved in RNA splicing and translation. Regulatory ncRNAs, including short-interfering (si) RNA, can play an important role in gene expression through both transcriptional and posttranscriptional mechanisms as well as through alteration of higher-order chromatin structure.
Advances in our knowledge have indicated that the concept of the “gene” as an essentialist biological entity with an unambiguous nature and clean boundaries is unsustainable. Genes are not discrete entities like atoms of gold and silver. They are dynamic parts of biological systems of immense complexity. The discovery of specific genes that are involved in the etiology of psychopathology will not likely prove to be the basis on which to build an essentialist and categorical model of psychiatric diagnosis.
Conclusions
Contrary to the widely cited work of Robins and Guze
(2), the familial aggregation of a single putative psychiatric syndrome provides at best quite limited evidence for the validity of that syndrome. Psychiatric genetics can supply useful information about the etiologic relationship between two disorders, although how that information is used in nosologic decisions (for example how it would be evaluated, compared to information on environmental risks or pharmacologic response) is outside of a strictly scientific domain. Whether molecular genetics will provide greater insights into our major diagnostic conundrums than has been obtained by more traditional genetic methods is far from certain. Evidence that one or a small number of individual genes or genomic regions impact on risk for two disorders is not likely to be nosologically definitive. Although essentialist gene models for psychiatric disorders are conceptually appealing, they are not well supported by available data. Indeed, such models may not apply even to more traditional Mendelian disorders. The hope that we will be able to develop categorical psychiatric diagnoses (i.e., “carving nature at its joints”) solely as a result of gene discovery is implausible; the genes found to date for psychiatric illness have far too small an effect size. (However, as has proven to be the case in Alzheimer’s disease
(41), it is possible that multiple genes will together point to a particular pathophysiological pathway that may have more explanatory power than the individual genes themselves). The project to ground our messy psychiatric categories in genes—as an archetypal natural kind—may be in fundamental trouble as advancing research suggests that the very concept of “the gene” as a discrete entity is itself more and more in doubt.
Psychiatric genetics has in the past and likely will continue in the future to provide important insights into the etiology of psychiatric and substance use disorders. These developments—particularly those involving molecular genetics—have, however, raised expectations that such advances will also produce major breakthroughs in psychiatric nosology. In this essay, I have reviewed these claims and have come to a largely skeptical conclusion.