Parental PTSD appears to be a salient risk factor for the development of PTSD, as evidenced by a greater prevalence of PTSD, but not trauma exposure, in adult offspring of Holocaust survivors with PTSD relative to those without the disorder
(1) . In addition, significantly lower 24-hour mean urinary cortisol excretion has been reported in offspring of Holocaust survivors with PTSD than in offspring of Holocaust survivors without PTSD
(2) . Other studies have reported negative correlations between cortisol levels and 1) parental PTSD symptom severity in adult offspring of Holocaust survivors (3) and 2) year-old infants born to mothers who were exposed to the attacks on 9/11 while pregnant (4).
That cortisol levels might be associated with some aspect of vulnerability to PTSD rather than symptom expression may explain the lack of consistency in observing low cortisol levels in studies of PTSD samples compared with other subject groups, since lower cortisol levels may only be present in trauma survivors showing specific risk factors (5). In contrast, the enhanced plasma cortisol suppression in response to low doses of dexamethasone appears to be a more robust correlate of PTSD, having now been reported to occur in each published report using this methodology (reviewed by Yehuda
[5] ). Accordingly, the present study examined the relationship between negative feedback inhibition, as measured using a low-dose dexamethasone suppression test, and the risk factor of parental PTSD to evaluate specifically the extent to which this measure may be associated with PTSD risk versus trauma-related experiences and symptoms in adult offspring.
Method
Participants
Twenty-one men and 20 women participated in the study. Recruitment was through advertisements requesting Jewish volunteers interested in research examining transgenerational effects of the Holocaust. Procedures were approved by the institutional review board at the Mount Sinai School of Medicine, and all participants provided written informed consent.
Holocaust offspring were born after 1945 and raised until age 18 by at least one biological parent who was interned in a Nazi concentration camp during World War II. This group was further subdivided on the basis of whether at least one parent had PTSD. Comparison subjects were Jewish and similar in age, but their parents had not been exposed to the Holocaust. Participants were excluded if they had psychotic or bipolar illness; alcohol or substance dependence; or major medical, endocrinological, or neurological illness confirmed by medical examination. Three participants had been stabilized with psychotropic medications.
Clinical Assessments
Information about traumatic life events was obtained using the Trauma History Questionnaire (6), and PTSD was diagnosed using the Clinician-Administered PTSD Scale (7). Other axis I diagnoses were determined with the Structured Clinical Interview for DSM-IV administered by trained psychologists or psychiatrists with established interrater reliability. Participants also completed the Civilian Mississippi Scale (8) and the Childhood Trauma Questionnaire (9).
Parental PTSD was assessed with the Parental PTSD Questionnaire, developed for rating parental PTSD symptoms by Holocaust offspring and validated based on consensus between offspring ratings and independent interviews of parents by clinicians (2, 10). Parental PTSD was determined on the basis of a positive endorsement of at least six symptoms distributed in the three symptom categories as required for a PTSD diagnosis by current criteria. Offspring of parents that were rated as having subthreshold symptoms (i.e., one symptom less than required in the avoidance or hyperarousal categories) were included in the nonparental PTSD group.
Biological Measures
Baseline blood samples were obtained by routine venipuncture at 8:00 a.m. The participant ingested 0.5 mg of dexamethasone at 11:00 p.m. that night, and blood samples for determination of cortisol and dexamethasone were obtained at 8:00 a.m. the following day.
Plasma cortisol and dexamethasone levels were determined by radioimmunoassay. The intra-assay and interassay coefficients of variation were 4.0% and 6.8%, respectively, for cortisol and 8.0% and 9.0% for dexamethasone.
Statistical Analyses
Natural logarithms of the raw cortisol data were used to minimize effects of outliers. Age, gender, body mass index (BMI), plasma dexamethasone levels, and medication status were tested for association with pre- and post-dexamethasone cortisol; the first four were used as covariates, since they were associated with either pre- or post-dexamethasone cortisol levels. Chi square and Fisher’s exact tests were used to test other differences among the three groups in categorical descriptors; analyses of covariance were performed to compare other continuous variables, followed by Dunnett T3 post hoc tests or by pairwise comparisons, as appropriate. To clarify the distinction between subject and parental symptoms, partial correlations were performed to assess whether differences in cortisol suppression were associated with the total number of lifetime parental PTSD symptoms after we controlled for subjects’ current PTSD symptoms (Civilian Mississippi Scale scores), presence or absence of lifetime PTSD, severity of childhood trauma, and dexamethasone levels.
Results
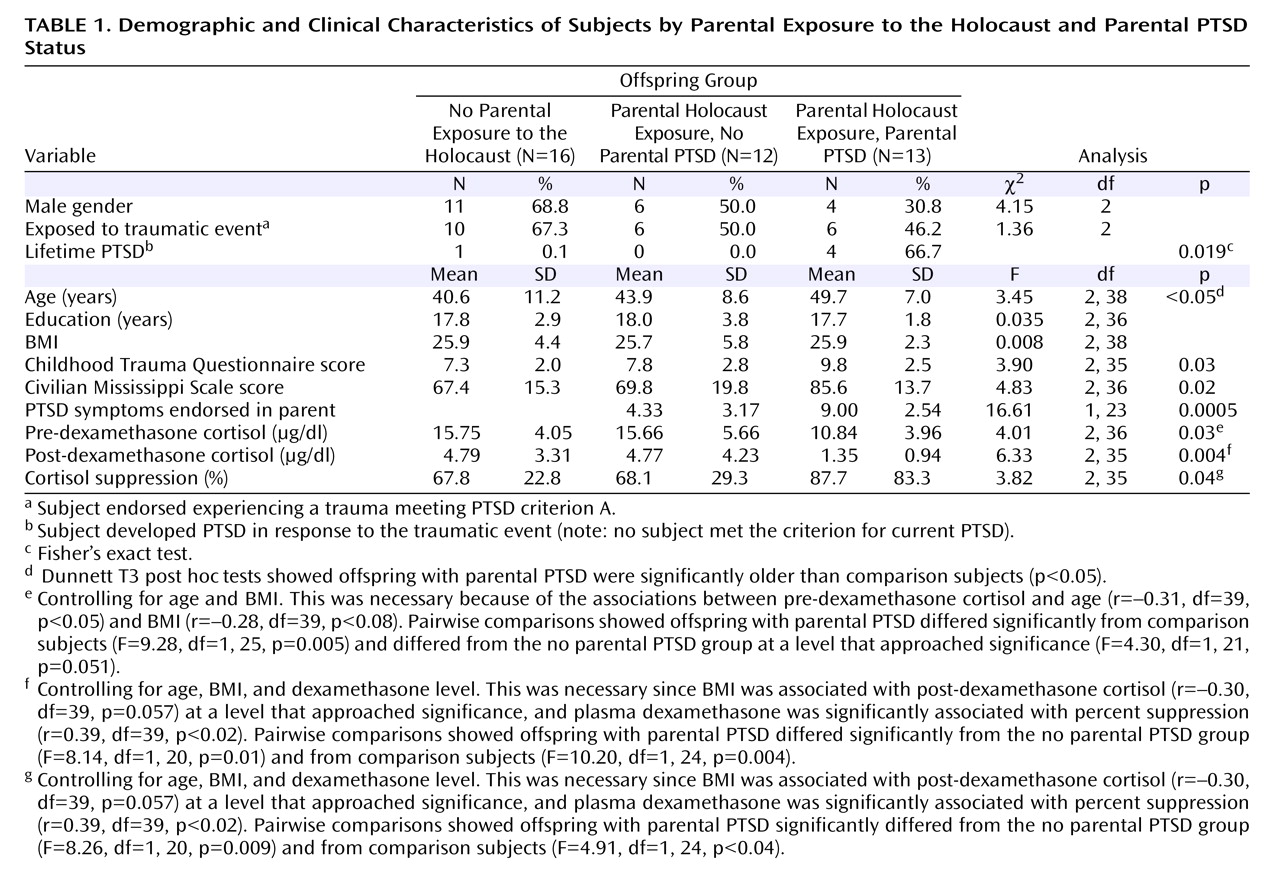
Table 1 shows that offspring of Holocaust survivors with PTSD were older than comparison subjects, but no group differences were noted in demographic variables or lifetime exposure to a traumatic event meeting PTSD criterion A. Offspring of Holocaust survivors with PTSD had higher Childhood Trauma Questionnaire and Civilian Mississippi Scale scores, and were significantly more likely to have past PTSD than the other two groups. No subject met criteria for current PTSD.
Significant group differences were also present in pre- and post-dexamethasone cortisol values. Repeated measures ANCOVA revealed a significant group-by-day interaction (F=3.66, df=2, 35, p<0.04), which was confirmed by pairwise comparisons that controlled for age, BMI, and dexamethasone levels to reflect greater cortisol suppression in offspring with parental PTSD than in offspring without parental PTSD (F=4.92, df=1, 20, p<0.04) or comparison subjects (F=5.56, df=1, 24, p<0.03), but not between offspring without parental PTSD and comparison subjects (p<0.54). There were no significant group differences in dexamethasone concentrations after age and BMI were controlled, nor was dexamethasone significantly associated with post-dexamethasone cortisol.
The total number of parental PTSD symptoms was significantly correlated with 1) severity of trauma-related symptoms (r=0.53, df=37, p=0.001), as rated on the Civilian Mississippi Scale; 2) childhood emotional abuse (r=0.33, df=36, p=0.041) and physical neglect (r=0.38, df=36, p<0.02), as measured by the Childhood Trauma Questionnaire; and 3) total Childhood Trauma Questionnaire score (r=0.41, df=36, p<0.02). Similarly, offspring Civilian Mississippi Scale scores were correlated with the childhood emotional abuse (r=0.55, df=36, p<0.0005), physical neglect (r=0.49, df=36, p=0.002), and total Childhood Trauma Questionnaire (r=0.52, df=36, p=0.001) scores.
It is important to note that the extent of parental PTSD symptoms was significantly associated with offspring post-dexamethasone cortisol after we controlled for offspring Civilian Mississippi Scale scores, presence or absence of lifetime PTSD, total Childhood Trauma Questionnaire scores, and dexamethasone levels (r=–0.35, df=32, p<0.05).
Discussion
The association between cortisol suppression and parental PTSD is noteworthy in that, unlike ambient mean 24-hour cortisol excretion rates, which are thought to constitute relatively stable measures, the cortisol response to dexamethasone has often been linked to more transient phenomena related to symptom severity and has been shown to normalize in successfully treated depressed patients (11). Examination of high-risk probands with a positive family history for depression have demonstrated alterations in cortisol negative feedback inhibition only when the high-risk subjects became affected, and persisted even after successful treatment (12). Thus, there may be an acquired component to the dexamethasone suppression test response that depends on an interaction of specific risk factors and environmental events.
Indeed, there is ample reason to suspect a role for environmental events in modulating the effects of parental PTSD, since offspring of parents with PTSD report significantly more emotional abuse and neglect than offspring of parents without PTSD and demographically comparable healthy subjects (5). Nonetheless, in the current study, the association between cortisol suppression and parental PTSD persisted after we controlled for childhood trauma and offspring’s own PTSD. Alternatively, alterations in glucocorticoid receptor sensitivity occurring very early in development, possibly even related to in utero factors, may be responsible for the findings. It is now well established that the activity of genes regulating HPA activity can be programmed by pre- and postnatal early life events, and even by differences in maternal care (13). Such influences in rat pups have been demonstrated to result in permanent changes in hippocampal glucocorticoid receptor expression and HPA function that are transmitted intergenerationally (14) and provide a clear molecular link between early environment and gene expression and function. It is interesting that the alterations observed are in the same direction as those described in PTSD (i.e., increased glucocorticoid receptor sensitivity, enhanced cortisol response to dexamethasone, lower ambient cortisol levels), offering proof of the principle that environmental exposures can result in the kind of changes that alter glucocorticoid receptor expression, and form the basis for individual differences in endocrine function including, perhaps, vulnerability to PTSD. One of the major limitations of studying endocrine aspects of PTSD has been the inability to know for certain whether what is being measured constitutes a change associated with PTSD pathophysiology, the trauma that produced it, or an earlier risk factor. To the extent that neuroendocrine measures associated with PTSD, or risk for PTSD, are somewhat stable, it is possible that they reflect even more enduring patterns (e.g., changes in patterns of DNA methylation) that reflect earlier life events, rather than cumulative effects of stress, and result in an increased cortisol signaling capacity so that lower levels of cortisol efficiently suppress HPA function. Under certain environmental conditions, such as in response to a traumatic event, increased cortisol signaling might alter or interact with psychological and biologic risk factors to result in the PTSD clinical phenotype. The current findings constitute support for employing a transgenerational approach to the study of biologic risk factors in PTSD.