Converging evidence in the last decades have suggested an organic basis for the pathogenesis of obsessive-compulsive disorder (OCD), an anxiety disorder characterized by distressful inability to suppress repetitive, intrusive thoughts and performance of repetitive actions interfering with home, school, work, and interpersonal functioning. Clinical, neuroimaging, and neurophysiological findings in OCD have pointed to dysfunction of orbitofrontal cortex, anterior cingulate, basal ganglia, and other subcortical limbic structures
(1), some of which are also involved in motor control. Motor cortical activation during motor preparation and execution is related to decreased expression (event-related desynchronization)
(2) of alpha and beta EEG sensorimotor rhythms. Increased expression (event-related synchronization) of the sensorimotor rhythms occurring after movement termination is considered a sign of cortical idling
(2). Moreover, postmovement beta synchronization corresponds in time to corticospinal inhibition
(3). The event-related desynchronization/synchronization pattern is abnormal in Parkinson’s disease and is characterized by dysfunction of basal ganglia and of cortical circuits related to motor programming
(4,
5). We investigated involvement of motor control in OCD by analyzing event-related desynchronization/synchronization.
Method
Subjects included 10 right-handed patients diagnosed with OCD according to the DSM-IV criteria (seven females; mean age=28.1 years, SD=7.2; disease duration=6–28 years; scores on the Yale-Brown Obsessive Compulsive Scale
[6]: mean total score=28.5, SD=4.8, range=23–37; mean obsessions subscale score=14.3, SD=3.2, range=10–20; and mean compulsions subscale score=14.2, SD=1.9, range=12–18). Exclusion criteria were a history of focal neurological disorders, systemic illness, head trauma, treatment with electroconvulsive therapy, drug abuse or dependence, or medication in the previous 3 weeks. Neurophysiological data were compared with those from a group of 10 healthy subjects (eight females; mean age=27 years, SD=4.7), all but one of whom were age-matched with the OCD subjects. The protocol was approved by the local ethics committee. After complete description of the study to the subjects, written informed consent was obtained.
Computerized 29-channel scalp EEGs with binaural reference, with electro-oculograph and bilateral electromyograph (EMG) monitoring of the extensor pollicis brevis, were recorded (band-pass DC to 50 Hz; 250 Hz sampling frequency). Subjects performed about 80 brisk self-paced extensions of the right thumb (one every 7–10 seconds) while sitting in an armchair, with their eyes open and their hands pronated.
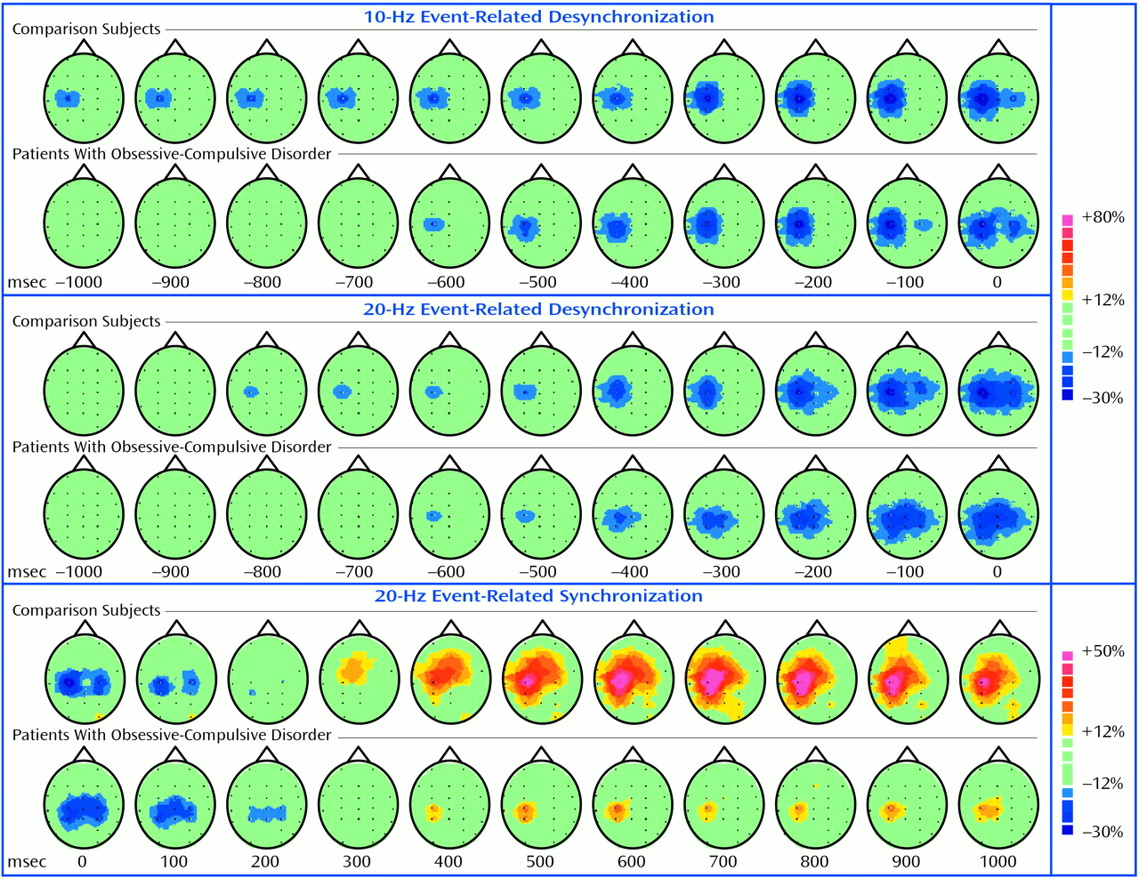
Event-related desynchronization/synchronization of the mu and beta (18–22 Hz) were then calculated
(7) over successive 50 msec time intervals between –2 sec and 1 sec both with respect to EMG onset and to EMG offset as the percentage of the average absolute amplitude of the reference period (between –2.5 and –2 sec before EMG onset). Trials with artifacts or behavioral errors in the EMG were not analyzed. The frequency band for the mu rhythm was individually selected as the 3-Hz interval around the frequency displaying the highest power decrease in a 1-second epoch centered on movement onset. Group statistical analysis was performed by using two-tailed Student’s t tests for nonpaired data. The onset latency of premovement mu and beta event-related desynchronization and of postmovement beta synchronization (with respect to EMG onset and offset, respectively) were evaluated with the C3 electrode (overlying the contralateral hand sensorimotor area). The magnitude of mu and beta event-related desynchronization and of beta synchronization were considered in the 0–50 msec period after movement onset and in the 700–750 msec period after EMG offset, respectively.
Discussion
We found delayed onset of premovement mu event-related desynchronization and a lower amount of postmovement beta synchronization in OCD patients than in normal comparison subjects. The lack of significant group differences in beta event-related desynchronization onset may be explained by a later onset of beta compared to mu event-related desynchronization even in normal subjects. Delayed event-related desynchronization of the mu rhythm has also been found in Parkinson’s disease
(4,
5), a basal ganglia disorder. Our finding of delayed mu event-related desynchronization in OCD patients is consistent with converging evidence indicating involvement of basal ganglia in this disease
(1).
A lower level of postmovement beta synchronization, which we found in OCD patients, has also been reported in Parkinson’s disease
(8) and may be interpreted as reflecting impairment of deactivation mechanisms after movement termination, since postmovement beta rebound corresponds in time to corticospinal inhibition
(3). A lower level of synchronization in OCD patients after a simple, self-paced movement may extend the concept of reduced inhibition in this disease and raises the question of whether this finding reflects the inability of OCD patients to refrain from performing impelling actions. Impaired inhibitory mechanisms in OCD have also been suggested by findings of event-related potentials with lower P300 amplitude in orbitofrontal areas of OCD patients during the “No Go” trials of the Go–No Go paradigm
(9). Moreover, reduced motor cortical inhibition in OCD patients has been found with transcranial magnetic stimulation
(10).
Further studies are needed to assess the anatomofunctional specificity of each event-related desynchronization/synchronization feature. Taken together, our findings support the view that brain structures involved in motor control are impaired in OCD.