Depending on the particular landmark discovery designated, we are now approximately half a century into the biological revolution in psychiatry, one that ushered in the generally held belief that psychiatric disorders cannot be fully understood outside of the context of biology. This revolution has greatly shaped the content of a journal such as this, filling its pages with reports concerning neurotransmitters, hormones, receptors, and both structural and functional neuroanatomy. It also has given rise to tremendous interest in genetic makeup as a predisposing factor toward psychiatric disorders.
There has been an explosion of interest in such gene therapy, giving rise to a number of specialized journals (e.g., Gene Therapy, Human Gene Therapy) and societies. Within the realm of neuroscience, work to date has focused on the application of gene therapy to neurological disorders, namely the slow neurodegenerative diseases and the acute necrotic neurological insults. There has been considerable progress in that arena, with a large preclinical literature now extant and the first clinical trials taking place.
It is that progress that motivates the present review. Understanding the reductive bases of psychiatric disorders is a far more subtle and difficult challenge than understanding the reductive bases of neurological ones. This review considers whether gene therapy in the nervous system has matured to the point at which it can start to be brought to bear on psychiatric disorders. Although gene therapy is unlikely to be applicable to psychiatry in clinical settings for some time to come, it is hoped that this review will demonstrate that the approach can be quite useful in preclinical studies.
Gene Therapy in the Nervous System
In the broadest sense, the goal in gene therapy is to transfer genes in that will have some salutary effect on cellular function. This review will shortly discuss what form such interventions have taken in the neurological realm and what form they might take in the psychiatric one, and it is worth initially discussing features of gene therapy in the nervous system, independent of the actual genes transferred.
The predominant challenge in this field is to find effective vectors for transferring such genes into the nervous system. The reasons for this being challenging are numerous. First and most important, neurons in the adult brain are primarily postmitotic (defined in
Appendix 1), and some of the most effective gene therapy vectors (for example, modified retroviruses) work by integrating their transgenes into the host DNA during cell division (reviewed in reference
1). Thus, one is quite limited in the range of vectors available. Second, the brain is far less accessible than most other organs, making for difficulties in delivering vector in sufficient quantities. Next, as attested to by the size of neuroanatomy texts, the brain is immensely heterogeneous. One consequence of this is a greater demand for anatomic accuracy in the delivery of vectors. A second, less obvious difficulty is that certain gene transfer vectors more readily infect certain neuron types than others
(2). Finally, because of their size, elongated morphology, and atypically high metabolic demands, neurons are fragile cells. As a result, some gene transfer techniques (e.g., electroporation or high-pressure injection of DNA with “gene guns”) are more likely to damage neurons than other cell types
(3).
Because of these issues, most gene therapy studies in the nervous system have made use of one of three approaches. In the first (
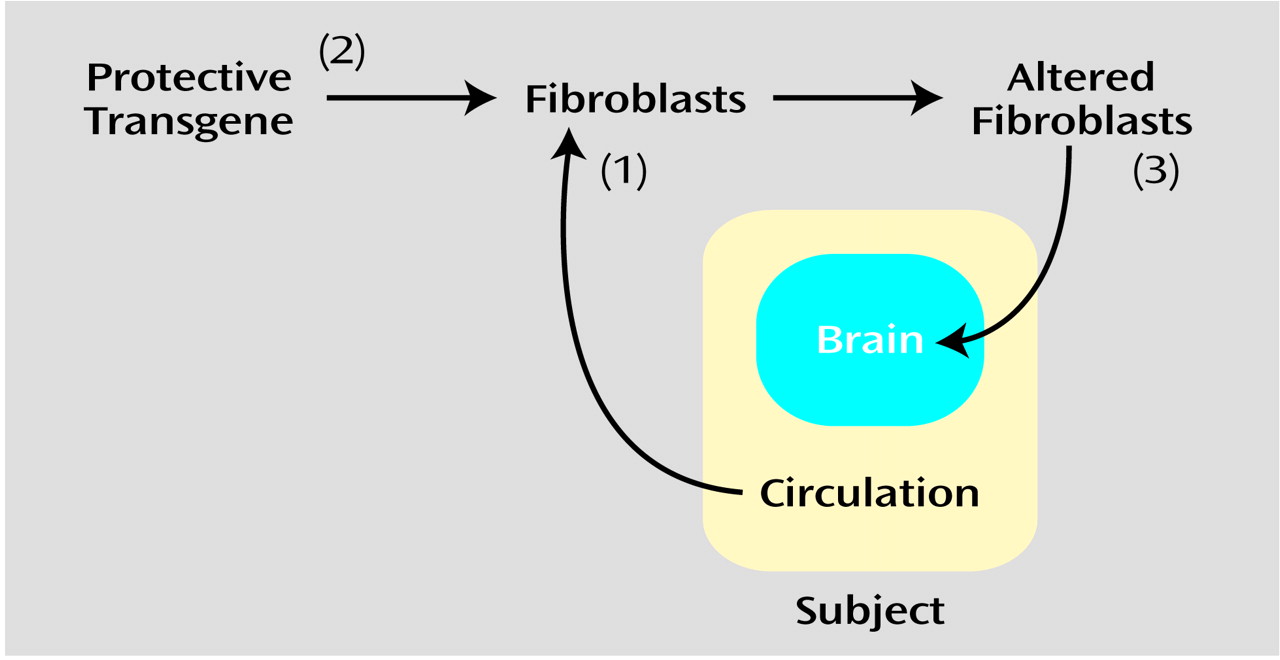
Figure 1), cells (typically fibroblasts) can be removed from the body, genetically modified ex vivo with a retrovirus, and then transplanted into the nervous system (compare with reference
4). As an example of this approach, a first gene therapy trial is currently being conducted in which fibroblasts engineered to secrete growth factors have been implanted into the brains of Alzheimer’s patients (Tuscinski, personal communication, 2002).
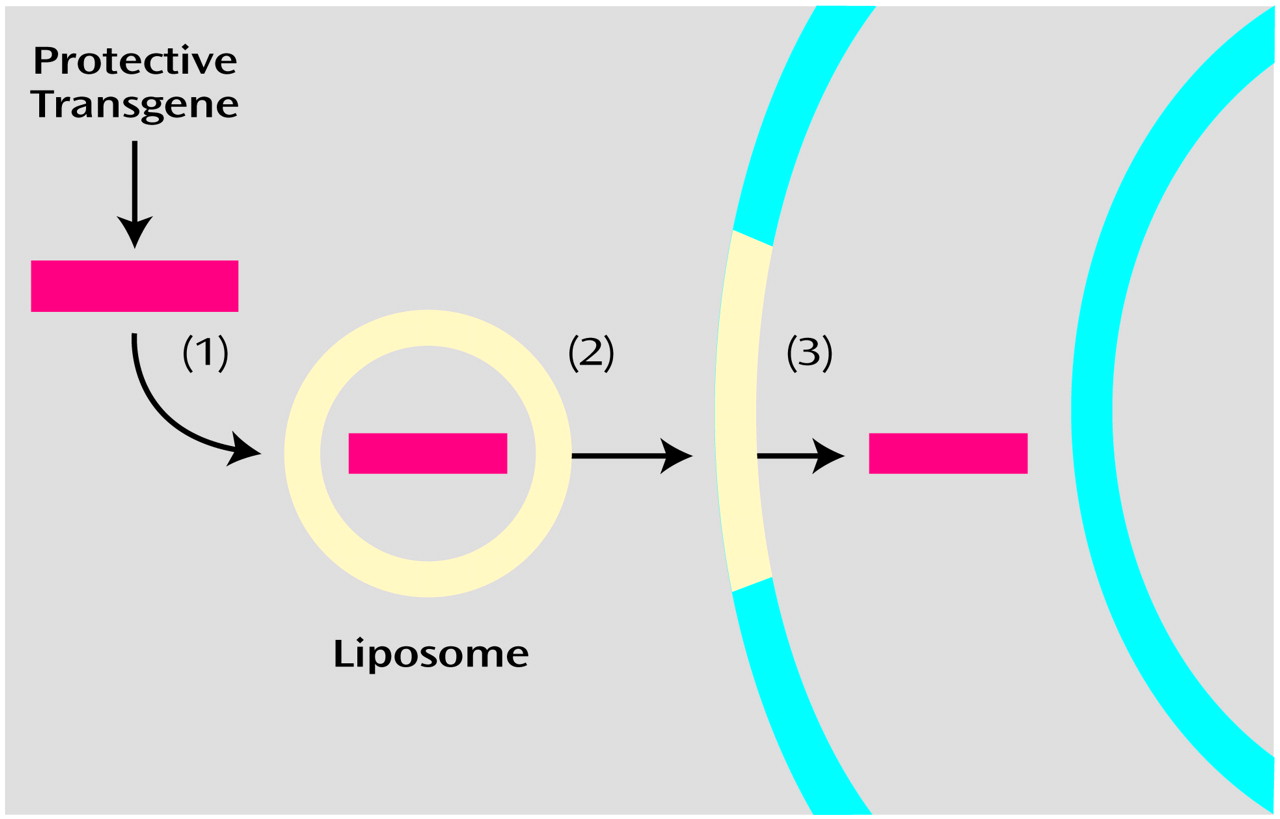
A second approach (
Figure 2) is to incorporate DNA into liposomes, whose lipid-rich coat can readily merge with cell membranes, allowing entry of the DNA (e.g., reference
5). In elaborations on this, the liposomal coat can be formed with agents that actively promote fusion with cell membranes, or it can be formed with ligands for cell surface receptors, facilitating endocytosis (defined in
Appendix 1) of the DNA (e.g., reference
6).
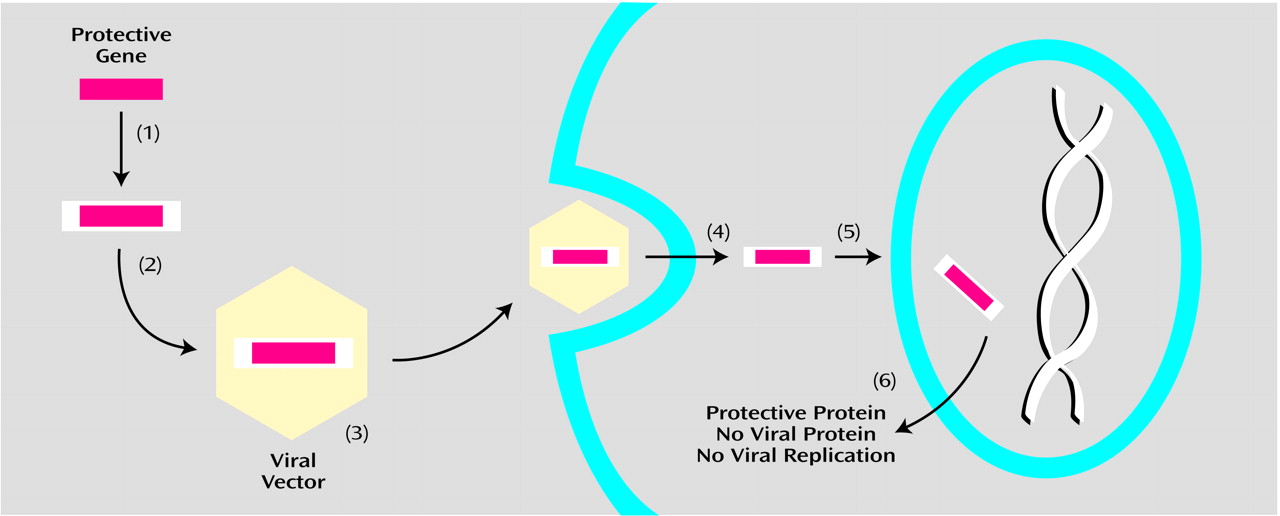
The third approach (
Figure 3) uses vectors derived from neurotropic viruses (defined in
Appendix 1) that are able to infect postmitotic cells. These include herpes simplex virus 1, adenovirus, adeno-associated virus, and lentiviruses. The basic strategy with such viruses is to 1) remove endogenous viral genes that could lead to damaging replication, 2) retain viral genes essential for infection and viral gene transcription, and 3) insert novel transgenic DNA.
The use of viral vectors has been the more common technique in the field and merits some review. There is no obvious virus that constitutes the ideal. Instead, viruses differ in their advantages and drawbacks. Virally derived vectors differ as to how much DNA they can transfer (with herpes simplex virus 1 having the largest capacity), duration of expression (with lentiviruses producing the longest and herpes simplex virus 1 the shortest), how quickly they express (with herpes simplex virus 1 being the most rapid), and how much inflammation they cause (with early versions of adenoviral vectors being particularly problematic). In addition, thanks to a detailed understanding of the functioning of many genes in these viruses, hybrid vectors have been constructed. This allows one to combine the advantageous traits of different viral vectors. For example, herpes simplex virus 1/adenovirus hybrids have been constructed that combine the long-term expression of the latter with the DNA packaging capacity of the former
(7).
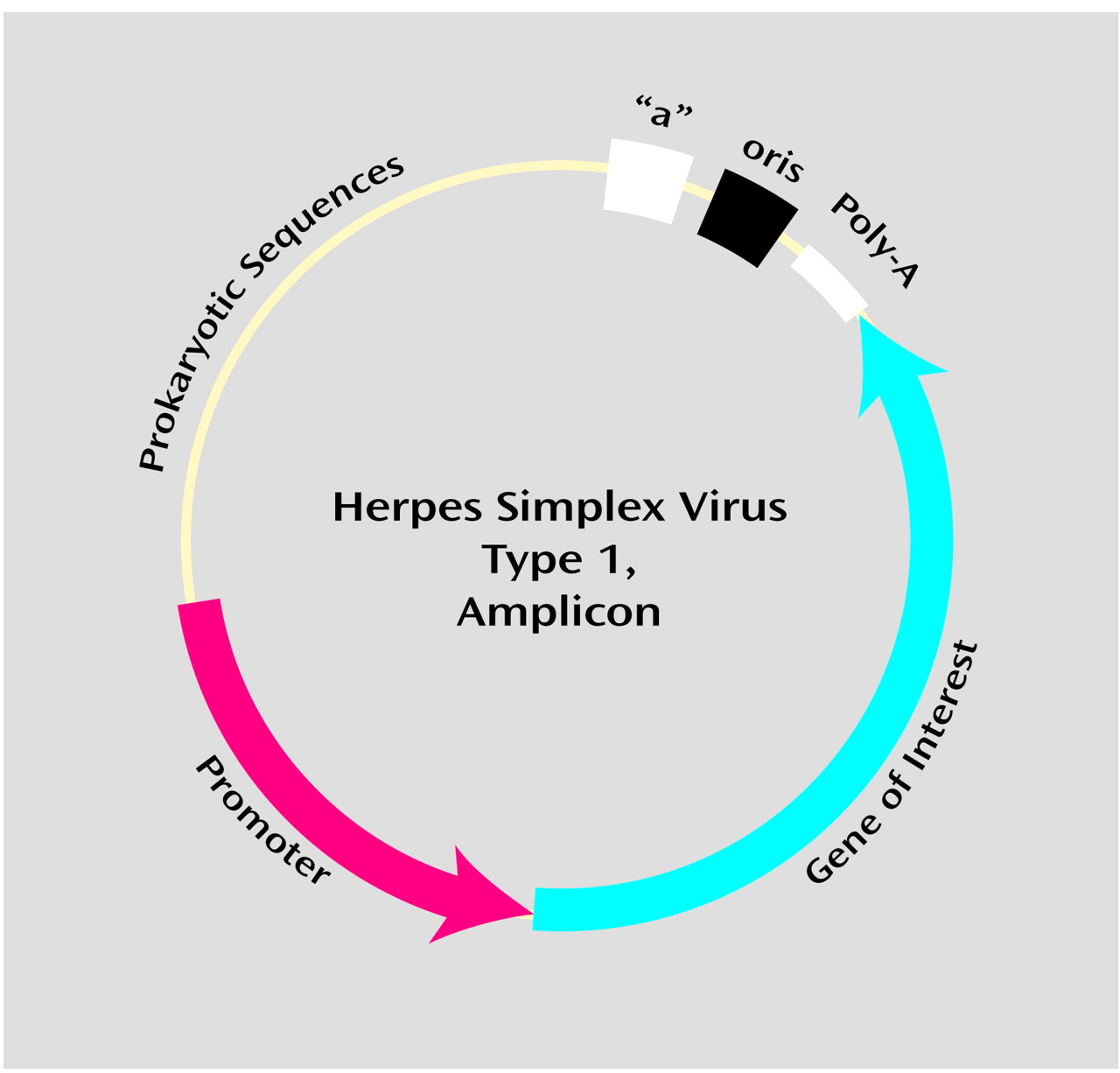
Even within a single viral class, there are options as to the type of vector used. For example, vectors derived from herpes simplex virus 1 can be of the recombinant type, a top-down approach in which recombination (defined in
Appendix 1) is used to strip herpes simplex virus 1 of the minimal number of genes needed to make the vector safe and incapable of replication. In contrast, herpes simplex virus 1 vectors can also be of the amplicon type, a bottom-up approach in which the minimal number of herpes simplex virus 1 genes needed for infection and transcription are added to a benign bacterial plasmid (defined in
Appendix 1), which is then packaged into a herpes simplex virus 1 viral shell (
Figure 4). These two types of vectors differ in their potency of expression, their cytopathicity (with a basic trade-off between the two), and their ease of generation
(8). As another example, versions of adenoviral vectors have been generated that differ as to the number of endogenous viral genes deleted; “gutless” adenoviral vectors represent the current extreme of truncating the viral genome
(9). These modifications have been prompted by the inflammatory tendency of adenovirus.
Once generated, vectors can be delivered to target neurons through a number of routes. The most common in preclinical studies has been microinfusion of the vector directly into brain tissue, from whence the vector infects nearby neurons and glia (with viral vectors differing in their preference for one cell type over the other) (e.g., reference
10). While this allows very local targeting, the drawbacks are that this requires stereotaxic surgery and that vectors spread only minimal distances from injection sites. Another route is to infuse vectors into the ventricles. The obvious advantages here are easier and more diffuse delivery. As a drawback, the vector does not penetrate far from the ventricles, limiting the usefulness of this approach for distant brain regions. Even more important, in this approach, it is typically the ependymal cells bordering the ventricles that are infected rather than the neurons. Thus, one is restricted to delivering transgenes whose protein products are secreted and work extracellularly rather than within neurons. This approach has been successful in overexpressing some anti-inflammatory agents to decrease neurotoxicity after necrotic insults
(11,
12). To date, there have been few successes with what would initially seem to be a far easier approach of delivering vector by means of the circulation because of the difficulties in vectors passing through the blood-brain barrier. In a recent exception
(13), the surfaces of liposomes were modified to contain antibodies against the transferrin receptor. The liposome was shown to then interact with such receptors, found on endothelial cells of the blood-brain barrier, triggering receptor-mediated transfer of the vectors across the barrier.
These represent some of the most common techniques for introducing novel DNA into the nervous system. As a promising elaboration on them, there have been reports of expressing multiple transgenes. This can be accomplished by infecting with multiple vectors (e.g., reference
14) or with a vector expressing multiple transgenes (e.g., reference
15). As an example of the potential benefits of this ambitious approach, in a rodent model of Parkinson’s disease, overexpression of both a growth factor and an inhibitor of programmed cell death was more protective than either alone
(16).
One problem is that viral vectors infect only a subset of neurons and glia in the field of spread from their site of delivery. Thus, one must often develop means for documenting which cells have been infected in order to document the efficacy of some therapeutic intervention. A solution is to construct vectors that, in addition to expressing the transgene under consideration, also express a “reporter gene”—one whose protein product does not alter cellular function but can be used as a marker of successful expression. Such reporter genes include the
E. coli gene beta-galactosidase, whose protein product produces a blue signal in the presence of a particular chromogenic substrate (
Figure 5). Other reporter genes express proteins that fluoresce a particular color at a certain wavelength; thus, exposing a tissue section to that wavelength of light will reveal which cells have been successfully infected. One challenge in the realm of reporter genes is to find one that is not so large that it precludes the inclusion of the transgene under study. A second is to ensure that expression of the reporter and transgene are coupled tightly enough so that the detectable expression of the former is truly reliable as an indicator of expression of the latter. One solution to that is to construct a gene that makes a fusion protein, a combination of the transgene under study with a tail consisting of a reporter protein
(17). This ensures 100% covariance between transgene and reporter; as a drawback, one must then initially document that the fusion protein functions as does the original transgene (which is difficult to answer when the function of the transgene is the rationale for the study in the first place).
Reporter genes are also essential in constructing appropriate controls for gene therapy studies. No matter how benign and minimal a viral vector is used, there is still the potential that viral infection itself, independent of the transgene expressed, will alter function of the host cell in some significant manner. Thus, the most typical control used in the field has become a vector expressing the same reporter gene but without the transgene in question; in that circumstance, care must be taken to ensure that roughly equal titers of the experimental and control vector are used.
A measure of the importance of reporter genes is seen in studies using gene therapy strategies to decrease neuron loss after experimental infarct due to occlusion of the middle cerebral artery. This produces a large field of damage in the region of the striatum, and few gene therapeutic interventions have even come close to being sufficiently successful as to decrease overall infarct volume. This is because the volume of tissue infected by viral vectors is vastly less than the volume of the infarct. Thus, rather than asking whether a particular intervention decreases infarct volume, the inclusion of a reporter gene allows one to instead ask whether more cells infected with the experimental vector survive the infarct than do cells infected with the control vector
(18).
This broad overview now allows us to consider the issue of which transgenes are expressed. More technical reviews of vector issues in neuronal gene therapy are available elsewhere
(1,
2,
8,
19).
Gene Therapy Against Neurological Insults
As noted, there have been some striking preclinical successes in gene therapy against neurological diseases and insults, and they are worth reviewing as a prelude to considering gene therapy in the more difficult realm of psychiatric disorders.
Gene therapy against a neurological insult can be conceptualized as accomplishing one of two things. The first would be to enhance a cellular process that increases the likelihood of neuronal survival. One version of this would be to replace an endogenous factor that is lost for congenital or environmental reasons. An archetypal example of that would be gene therapeutic replacement of a missing enzyme in a lipid storage disease, as shown in a mouse model of Fabry disease
(20). Another version would be to boost some endogenous process. Neurons are not passively assailed by neurological insults; instead, there is an array of neuronal defenses that are mobilized in response to injury
(21), and an approach can be to identify a key defense and attempt to bolster it. As an example, after both ischemic and excitotoxic insults, neurons up-regulate the expression and activity of various antioxidant enzymes in an attempt to contain the oxygen radicals being generated (e.g., references
22,
23). As such, one possible intervention is to use a gene therapy approach to further overexpress those antioxidants
(24,
25). In addition to the enhancement approaches of overexpressing “more of what is there” or “more of what would normally be there,” one can enhance function by creating protective pathways that are completely novel to the mammalian nervous system. As a speculative example, extremophiles are species, typically invertebrates, that live at extremes of temperature, oxygen pressure, pH, and so on and presumably have evolved genomic adaptations for dealing with such environmental challenges. This raises the potential of then harnessing such mechanisms for dealing with, for example, the extremely low oxygen pressure of a hypoxic-ischemic insult to a mammalian neuron.
Conceptually, the other broad gene therapeutic approach for neurological insults is to inhibit a cellular process that helps mediate damage. This requires insight as to the molecules that mediate damaging events, such as oxygen radical generation, cytoskeletal degradation, or the DNA fragmentation of programmed cell death. One therapeutic strategy is to express a protein that inhibits such a process. For example, a number of studies have shown neuroprotection with overexpression of inhibitors of programmed cell death (e.g., references
26,
27). A second approach is to follow an antisense strategy, expressing a stretch of mRNA that is antisense to that coding for an endogenous, endangering factor. The two then combine, leading to unnaturally double-stranded RNA, blocking protein translation (e.g., reference
28).
With these varied approaches, there have now been numerous reports of protection against neurological insults with gene therapy strategies. A number of studies have also targeted the slow neurodegenerative insults. Best studied have been models of Parkinson’s disease in both rodents and nonhuman primates. These have reported sparing of neuron number, an enhancement of dopaminergic transmission, and/or maintenance of motor function with overexpression of neurotrophins, inhibitors of programmed cell death, or tyrosine hydroxylase (reviewed in references
29,
30).
A body of studies has targeted the necrotic neuron death that results from hypoxia-ischemia, seizure, or hypoglycemia. All have focused on the central cascade of events mediating such neuron death. As a first step, this cascade involves an excess of the excitatory neurotransmitter glutamate accumulating in the synapse, both because of excessive release into the synapse and energy-related failure of glutamate removal from the synapse. Next, such excessive glutamatergic excitation leads to a pathological accumulation of calcium in the cytosol of the postsynaptic neuron. This involves both an excess entry of calcium into the cytosolic pool and failure of the costly task of calcium sequestration and extrusion. This calcium excess leads to promiscuous overactivation of calcium-dependent processes, resulting in cytoskeletal degradation, protein misfolding, aggregation, and, as arguably the key pathogenic event, generation of oxygen radicals. These derangements lead to necrotic neuron death and secondary inflammatory injury in the region. In a small subset of neurons, oxygen radical accumulation serves as a trigger for an alternative death pathway, namely apoptosis, a subtype of programmed cell death that does not trigger inflammation (reviewed in reference
31).
Various transgenes delivered by means of viral vectors have been reported to protect against necrotic insults. Efficacious approaches have included overexpression of a glucose transporter in order to bolster neuronal and glial energetics during an insult, a calcium-binding protein to sequester excess calcium, and heat shock proteins to counter protein misfolding, antioxidant enzymes, apoptosis inhibitors, or anti-inflammatory proteins (reviewed in reference
32).
In these studies, the endpoint has typically been whether there is a decrease in lesion size and/or in the number of dead neurons. Some have investigated the biochemical steps mediating any such protection. It is worth reviewing one example of this as a model for what would need to be done in targeting a psychiatric disorder. One of the most detailed examples documented concerns overexpression of the Glut-1 glucose transporter. The rationale for its use was the fact that necrotic insults ultimately represent energy crises, either impairing the capacity of neurons to generate adequate energy (as in hypoxia-ischemia or hypoglycemia) or pathologically consuming energy (as in prolonged seizure)
(10). Moreover, as noted, key mediators of neuron death after necrotic insults involve energy depletion in that the costly tasks of high-affinity glutamate removal from the synapse or calcium removal from the cytosol fail. Thus, this prompted overexpression of a glucose transporter, and this strategy has been shown to protect against a variety of necrotic insults, both in vitro and in rodents in vivo (reviewed in reference
32). As a first, most proximal endpoint that must be demonstrated to make sense of such protection, one would need to show that its overexpression does indeed lead to higher levels of glucose uptake in infected cells. This has been shown both in vitro and in vivo
(33). A next step is the demonstration that this results in the buffering of energy stores (e.g., ATP levels) and cellular metabolism during an insult
(10,
34). Next, it is critical to demonstrate that this results in enhancement of energy-dependent neuronal defenses, such as glutamate uptake and calcium extrusion
(34). Given this sequence of findings, it is then not surprising to see that neuron death is also decreased. Such a stepwise approach is useful. If the intervening steps between overexpression of transgene and altered rates of neuron death are observed as anticipated, this serves as an internal control for our knowledge of the underlying biology. And in contrast, if something unanticipated is observed (for example, if overexpression of a glucose transporter were found to decrease neuron death but not to alter glucose transport), this may be of considerable heuristic value.
Two additional steps are needed in documenting the efficacy of a certain gene therapy, such as the overexpression of the glucose transporter. In most cases, necrotic neurological insults occur without warning, meaning that gene therapy (in whatever incarnation might be of clinical use) would often need to be initiated after the insult rather than in anticipation of it. Thus, it becomes important to demonstrate whether there is a postinsult window of opportunity in which overexpression of the glucose transporter is still protective. For rodent models of necrotic insults, it appears that the vector must be introduced within a few hours (compare with reference
18). As the second step, little is gained if a neuron, spared from death caused by a necrotic insult, nonetheless is so damaged as to fail to function. Thus, it is critical to demonstrate sparing of function. This has been shown on both the behavioral and electrophysiological levels with overexpression of the glucose transporter, in contrast to some gene therapy strategies that have targeted later steps in this cascade of neuron death and failed to spare function
(35–
37).
From this brief overview of progress in the neurological realm, we can now consider application of these same approaches to psychiatric disorders.
Gene Therapy Against Psychiatric Disorders
A necrotic neurological insult, such as global ischemia, is the antithesis of subtlety. Life-or-death decisions are played out in vast numbers of neurons over the course of hours, driven by order-of-magnitude changes in rates of events (e.g., neurotransmitter release and reuptake) and in levels of key molecules (neurotransmitters, ions, and reactive oxygen species). Given the severity of this scenario, there can be a fair degree of tolerance of side effects of a particular gene therapy intervention. For example, overexpression of the calcium-binding protein calbindin D28K can be deleterious, disrupting synaptic plasticity in healthy hippocampal neurons
(38). Nonetheless, such transient disruption seems acceptable in the context of a massive necrotic insult, given the neuroprotective effects of transient overexpression of calbindin D28K against such an insult
(39–
41).
In contrast, the biology of a psychiatric disorder is intrinsically subtler than that of global ischemia, a grand mal seizure, or concussive head trauma. As a first question, can genes be transferred into enough neurons to actually alter a behavioral phenotype and can that alteration be detected amid the background of any side effects of the gene transfer?
This appears to be possible. As a first example, Martinez-Serrano and Bjorklund
(42) used the ex vivo gene transfer approach (defined in
Appendix 1) with immortalized neural progenitor cells engineered to overexpress and secrete nerve growth factor. Such cells were transplanted into the nucleus basalis and septum of middle-aged rats. Remarkably, this led to enhanced spatial learning in old age in these animals.
As a second example, aspects of addictive behavior have also been altered with gene transfer. One central feature of the neurobiology of addiction is the role of dopaminergic reward pathways. Overexpression of the dopamine signal transduction molecule CREB (by means of microinjection of a herpes simplex virus 1 vector into the nucleus accumbens) decreased the reward properties of cocaine
(43). Another more recent theme in addiction biology has been the role of synaptic plasticity in reward pathways in bringing about context-dependent addiction
(44). The neurotransmitter glutamate is central to such plasticity, and overexpression of glutamate receptor subunits can induce sensitization to morphine
(45).
A third example concerns reproductive behavior. Many rodent species, such as prairie voles, are monogamous, in which repeated mating between a male and a female brings about the formation of a stable pair bond between them. Vasopressin plays a key role in mediating the emergence of monogamy in males of such species. Recent fascinating work demonstrates that overexpression of the V1a vasopressin receptor in the ventral pallial region (by means of an AAV vector) accelerates the emergence of such monogamy
(46) (readers will no doubt differ in their opinions as to whether this constitutes a case of gene transfer altering normative behavior or of gene therapy correcting pathology).
Thus, gene transfer techniques have reached the point of being capable of altering a behavioral phenotype. As we now consider psychiatric disorders, it initially seems rather straightforward to apply some of the approaches used against neurological insults. As a potential example of the strategy of enhancing some intrinsic process, one might overexpress a benzodiazepine receptor to ameliorate anxiety. As a potential example of the inhibitory approach meant to counter some overactive intrinsic process, one might counter schizophrenia by expressing antisense against a dopamine receptor.
This is obviously vastly simplistic, and a key question is when would it be desirable to express a particular transgene. This ushers in the central point of this review. If one were forced to encapsulate the biology of psychiatric disorders in a single phrase, a plausible candidate would be “biological vulnerability.” From the first moment that those touched by psychiatric disorders assimilate a biological model involving genes, the critical concept that must be emphasized endlessly is that the biology of psychiatric disorders is not about inevitability but is instead about vulnerability and propensity. It is only in certain environments that the disease is likely to emerge.
Stated more abstractly, many psychiatric disorders have contingent qualities, experiential “if-then” clauses, explaining their emergence. As an obviously simplistic example, “
If you were abandoned by a critical loved one in your childhood,
then the likelihood of your concluding that no one will ever love you increases.” Or, “
If you are subjected to repeated aversive circumstances of lack of control,
then you become more likely to overgeneralize this to a global state of learned helplessness.” Or, “
If you are exposed to a major trauma in a circumstance that you had assumed to be safe,
then you become more likely to decide that there are never reliable safety signals and all circumstances demand a vigilant anxiety.” The actual if-then clauses involved seem to be more sophisticated than this, involving what neurobiologists term “coincidence detectors” of an “if A and B, then X” form. For example, significantly more predictive power is attained by switching from the simple if-then clause of, “
If you are exposed to a stressful life event,
then your risk of depression increases,” to the more complex, “
If you are exposed to a stressful life event
and you have a low sense of self-efficacy,
then your risk of depression increases”
(47).
If the biological predisposition toward a psychiatric disorder is more likely to emerge in certain circumstances, then gene therapy is more likely to be needed in certain circumstances. The bulk of viral vectors studied in the nervous system to date have been constitutive (defined in
Appendix 1) (which is to say that transcription commences immediately after infection). Is it possible to construct vector systems that are inducible (defined in
Appendix 1), whose expression is contingent upon the same events that increase the risk of psychiatric disease?
In beginning to understand how this can be the case, it is important to appreciate how the genome represents an informational system of if-then clauses. As is textbook knowledge, the vast majority of DNA does not code for genes that produce proteins. Instead, large proportions of nontranscribed DNA are made up of the promoter sequences (defined in
Appendix 1) that regulate when genes are transcribed. These form conditional if-then clauses of, “
If transcription factor (defined in
Appendix 1) A binds to promoter Y,
then transcribe gene Z,” or, more typically, more complex versions of, “
If and only if transcription factors A, B, and C all form a complex at promoter Y,
then transcribe gene Z.”
This regulatory organization allows gene transcription in the nucleus to be contingent upon events elsewhere in the cell. For example, cellular “stress” can take the form of misfolded proteins that are typically first sensed in the endoplasmic reticulum. There, their presence causes the cleavage of a transmembrane protein whose cytosolic fragment then translocates to the nucleus. There it acts as a transcription factor, binding to promoter sequences termed unfolded protein-response elements, leading to the synthesis of molecular chaperones that stabilize misfolded proteins
(48).
This organization also allows for gene transcription to be contingent upon events elsewhere in the body. This is most readily accomplished by means of hormones. In the case of peptide hormones, which bind to cell surface receptors, this typically involves ligand-induced activation of second messenger cascades, which lead to a change in the phosphorylation state and activity of particular transcription factors. With steroid hormones, which readily pass through lipid membranes and bind to intracellular receptors, the hormone/receptor complex itself acts as a transcription factor, binding to steroid-responsive elements (defined in
Appendix 1) within promoters
(49). Given this endocrine involvement, this regulatory organization also allows for gene transcription to be contingent upon events in the environment, outside the organism. As examples, a female rodent smelling the pheromones of a fertile male, a female elk hearing the long call of a roaring stag, and a male nonhuman primate seeing a female’s estral swelling all alter hormone secretory patterns and thus genomic events
(50).
Therefore, the likelihood with which the symptoms of a psychiatric disorder is expressed—and gene expression—can both be highly contingent upon environmental factors. Is it possible to construct vector systems that can be induced by the same external signals that increase the risk of a psychiatric disorder?
A number of regulated vector systems have been described in which the inducing signal is highly artificial, requiring intentional intervention. One early example is the tetracycline-responsive promoter system
(51). In a simplified explanation of this complex system, genes in
E. coli that are tetracycline resistant can be used to construct vectors where expression can either be induced or repressed by tetracycline administration
(52). Such a system has been applied in the nervous system
(53). In one interesting example of its use, neuroblastoma cells were engineered ex vivo to express beta-endorphin under the control of the tet system. When transplanted into the subarachnoid space, induction with tetracycline produced beta-endorphin secretion, alleviating pain
(54). The artificiality is obvious of having to administer tetracycline at the time of psychiatric risk, let alone the converse version of having to constantly administer tetracycline and withdraw it at the time of risk.
In another equally artificial approach, vectors have been constructed containing metallothionein promoters, inducible by the exogenous administration of heavy metals
(55) or promoters inducible by ionizing radiation
(56). In a clever variant on this approach, No and Evans
(57) constructed a vector expressing a fusion protein that combined a viral transcription factor with a receptor for the insect steroid hormone ecdysone. Thus, administration of ecdysone would cause the fusion receptor to translocate to the nucleus, where the viral transcription factor would activate transcription of a desired gene. The advantage here was that insofar as an insect steroid was completely novel to the rodent body, there could be no untoward side effects arising from transcriptional regulation of native rodent genes.
These prior vectors have conditional clauses of, “
If exogenous manipulation X occurs,
then induce gene Z.” Other regulated gene therapy systems have used a version closer to what is desired here, namely, “
If endogenous physiological event X occurs,
then induce gene Z.” As a first example, hypoxia induces transcriptional responses in the body that serve as defenses against this insult (for example, induction of genes relevant to angiogenesis, the growth of new blood vessels). This occurs by means of interaction with hypoxia-responsive elements (defined in
Appendix 1) in certain promoters. As a result, vectors have been constructed with hypoxia-responsive element promoters that are inducible by hypoxia
(58,
59). A number of studies have used glucose-regulated promoters. These have allowed the construction of vectors that express insulin in response to hyperglycemia, thereby alleviating the symptoms of experimental diabetes
(60,
61). The construction of vectors using prostate antigen promoters, rendering them androgen inducible, has also been reported
(55).
Therefore, gene therapy systems can be constructed that are inducible by events external to the infected cell, including systems that do not require exogenous manipulation by an experimenter. Is it possible to construct a vector with the conditional clause of, “If exogenous event X occurs, which increases the risk of a particular psychiatric impairment, then induce gene Z”? We have recently developed a system that may meet some of these specifications.
In considering the exogenous events that increase the risk for a psychiatric malady, arguably the best-documented example is the ability of stress to increase the likelihood of a depressive episode. The linkage between stress and depression is derived from a wide range of findings (reviewed in reference
62), including 1) epidemiological studies of predisposing factors toward depression in humans (with the recognition that after a sufficient number of such episodes, endogenous cycling of depression can occur), 2) the capacity for repeated stressors to induce a state of learned helplessness, 3) the ability of glucocorticoids, the adrenal steroids released during stress, to cause depression (as demonstrated with Cushingoid patients and individuals administered high-dose exogenous glucocorticoids), 4) the capacity for glucocorticoids to alter neurochemical events in a way that likely predisposes to depression, 5) the evidence that glucocorticoid synthesis inhibitors or glucocorticoid receptor antagonists can have antidepressive effects in some cases.
Thus, a prolonged excess of glucocorticoids can, as a first approximation, be a plausible signal of a greater risk of depression. As a steroid hormone, glucocorticoids, when complexed with their receptors, can act as transcriptional factors by means of binding to glucocorticoid-responsive elements
(63). As it turns out, herpes simplex virus 1 and other reactivating viruses (defined in
Appendix 1) have evolved the means to opportunize a glucocorticoid stress signal. As has been well documented, a variety of stressors, including social stress, can activate herpes simplex virus 1 out of latency
(64), the teleology being that the immune suppression that occurs during stress presents an advantageous time for the virus to replicate. As the mechanism to explain such reactivation, genes relevant to reactivation of herpes simplex virus 1 are under the control of a promoter containing a glucocorticoid-responsive element
(65).
Prompted by findings such as these, investigators have developed glucocorticoid- and stress-inducible vector systems, making use of promoters containing glucocorticoid-responsive elements. As one example, Ishii et al.
(66) did ex vivo gene therapy on a cell line engineered to secrete beta-endorphin under the control of a promoter containing a glucocorticoid-responsive element (continuing the theme of numerous types of viruses opportunizing glucocorticoid stress signals, the promoter in this case originated from mouse mammary tumor virus). Transplanting the cells into the subarachnoid space, they demonstrated that pharmacological levels of glucocorticoids could induce beta-endorphin release. Unfortunately, they did not test whether the stressor of pain itself could do so.
In another example, a glucocorticoid-inducible vector was constructed to contain the glucocorticoid-responsive element with mouse mammary tumor virus promoter that overexpressed growth hormone
(67). In the study most relevant to the current discussion, we constructed a family of herpes simplex virus 1 vectors containing promoters with five glucocorticoid-responsive elements
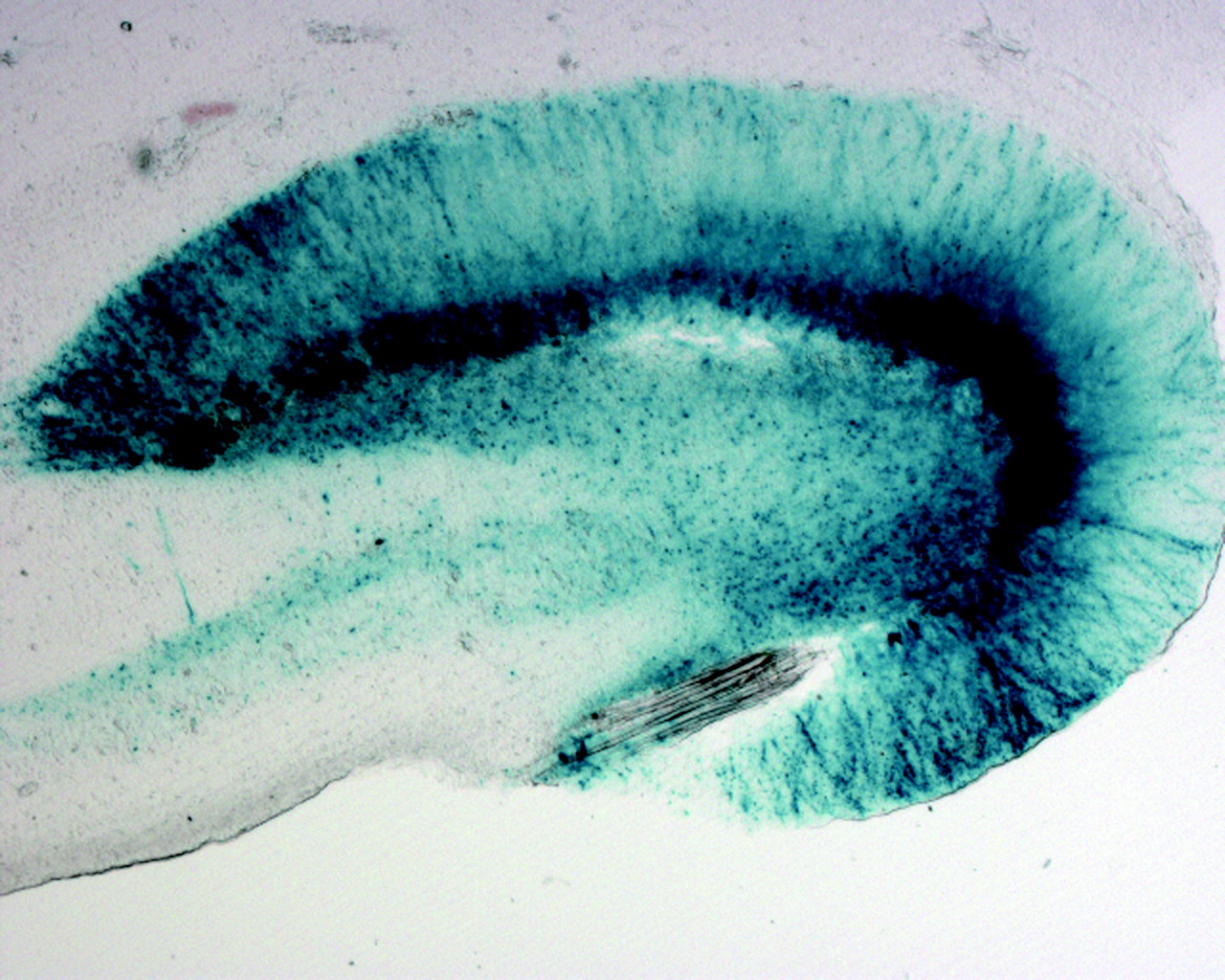
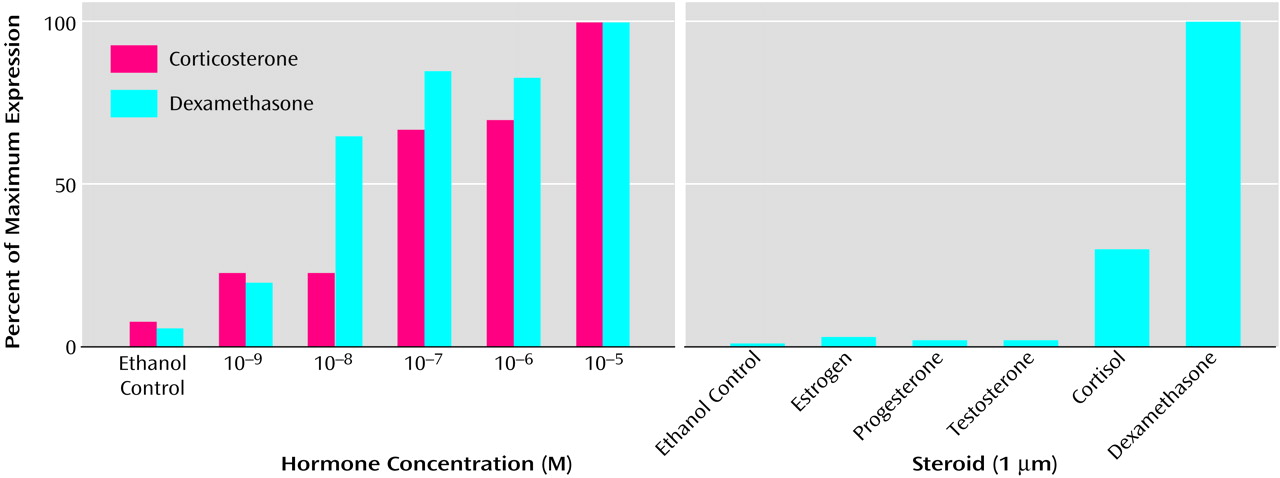
(68). Such vectors can be induced in hippocampal cultures and in the hippocampus in vivo by both endogenous and synthetic glucocorticoids in a dose-responsive manner but not by nonglucocorticoid steroids (
Figure 6). Moreover, these vectors can be introduced into neurons in a glucocorticoid-free environment, become transcriptionally quiescent, and then be induced by glucocorticoids at a later time. Most important, these vectors can be induced robustly by a stressor, namely an excitotoxic seizure, and, when expressing a protective transgene, can reduce subsequent neurotoxicity.
From the standpoint of gene therapy against a neurological insult, one can imagine any of a variety of protective transgenes being expressed. From the perspective of gene transfer, rather than gene therapy, we are currently exploring whether we can alter normative hippocampal function during stress with this vector system (specifically trying to reverse the capacity of stress to impair hippocampal-dependent cognition
[69]). And from the standpoint of psychiatric disorders, one can readily imagine a glucocorticoid-inducible vector overexpressing a gene thought to be relevant to the subsequent increased risk of depression, e.g., tryptophan hydroxylase or tyrosine hydroxylase.
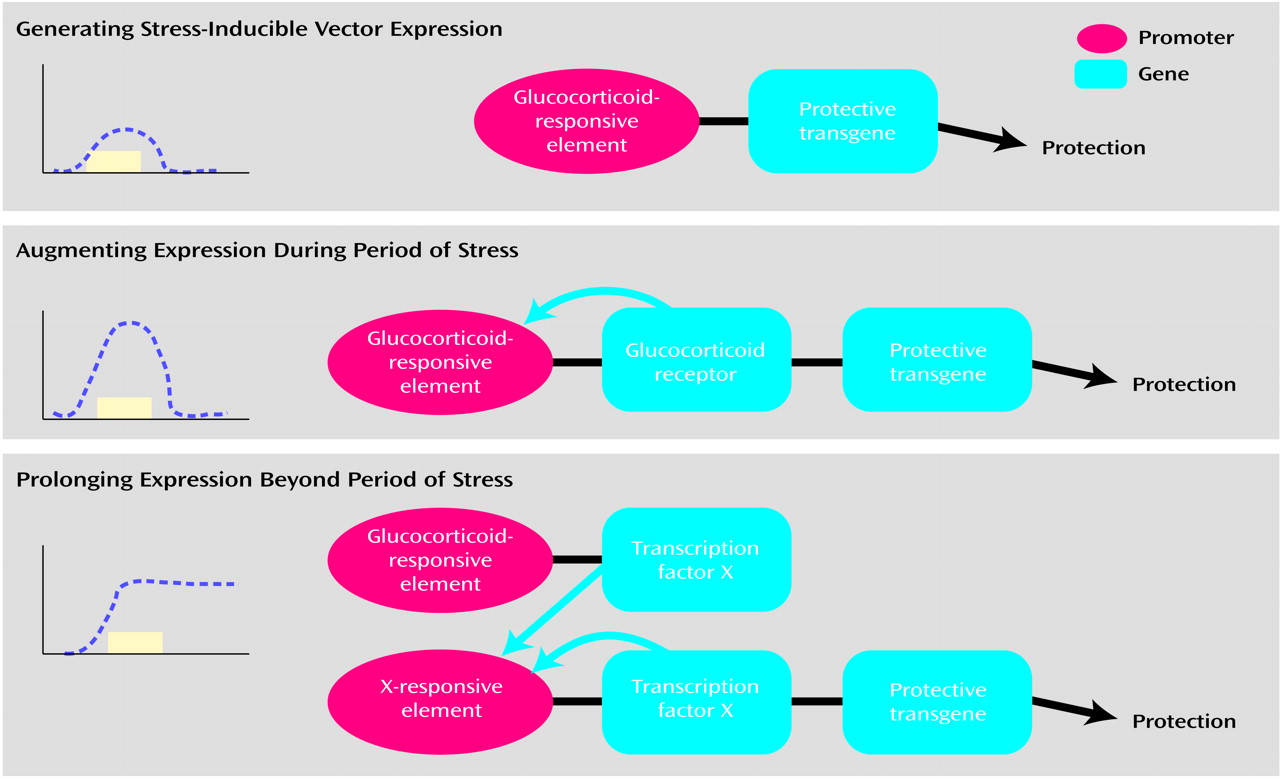
We are now exploring a number of elaborations on such a glucocorticoid-inducible system. In the version just described (
Figure 7, top, derived from reference
70), a transient stress signal, by means of glucocorticoid secretion, causes transcription of a protective transgene by means of a glucocorticoid-responsive element. A greater magnitude of expression can be attained with an amplification scheme (
Figure 7, middle). In this instance, the glucocorticoid-responsive element drives expression of two downstream genes (whose expression can be tightly coupled). The first expresses the glucocorticoid receptor. Such increased receptor levels generate a positive feedback loop, making the target cell more sensitive to the glucocorticoid signal. This leads to increased levels of the transgene (as well as the glucocorticoid receptor) as long as there continues to be a glucocorticoid signal. A third scheme allows for transgene expression beyond the period of glucocorticoid exposure (
Figure 7, bottom). This requires two constructs. In the first, a glucocorticoid-responsive element drives expression of transcription factor X. The second construct contains a response element to this transcription factor (i.e., an X-responsive element). This promoter drives the expression of both the protective transgene and of transcription factor X. This results in expression of the transgene beyond the period of glucocorticoid exposure.
Conditional gene therapy against a psychiatric disorder could take other guises, for example, with expression being regulated by pain, the presence of an abused substance, or numerous other possibilities. We are currently exploring another possible application. Considerable basic research suggests that anxiety and conditioned fear can arise from potentiation of synaptic plasticity in the subnuclei of the amygdala (compare with reference
71). We have constructed a number of vectors that express ion channels that hyperpolarize neurons, preventing such potentiation. The conditional nature of the expression of these transgenes is supplied by the fact that these vector systems are voltage dependent. We originally developed these with the goal of conditionally hyperpolarizing neurons undergoing epileptic seizures
(72). Theoretically, these could function to block amygdaloid plasticity during periods of the sort of hyperexcitation that might give rise to fear conditioning.