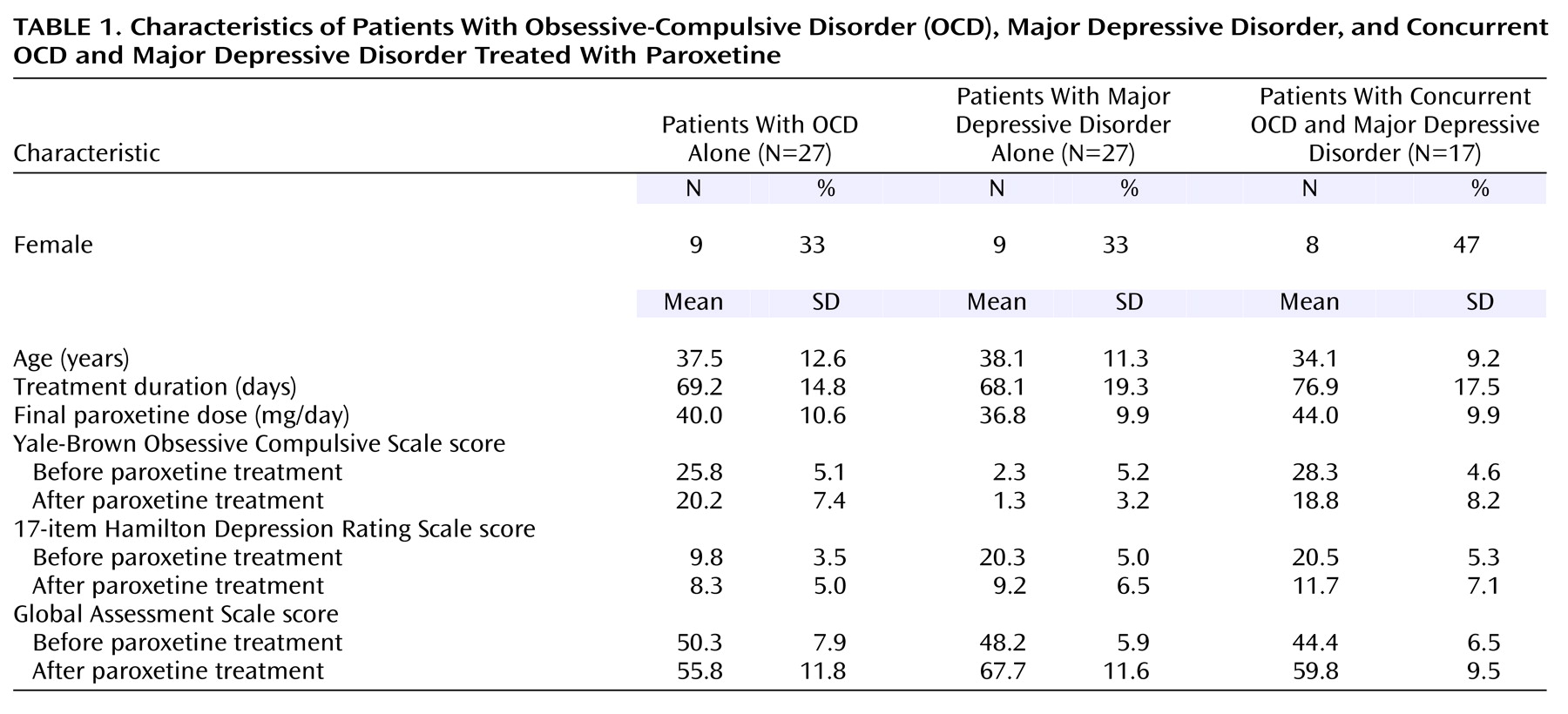
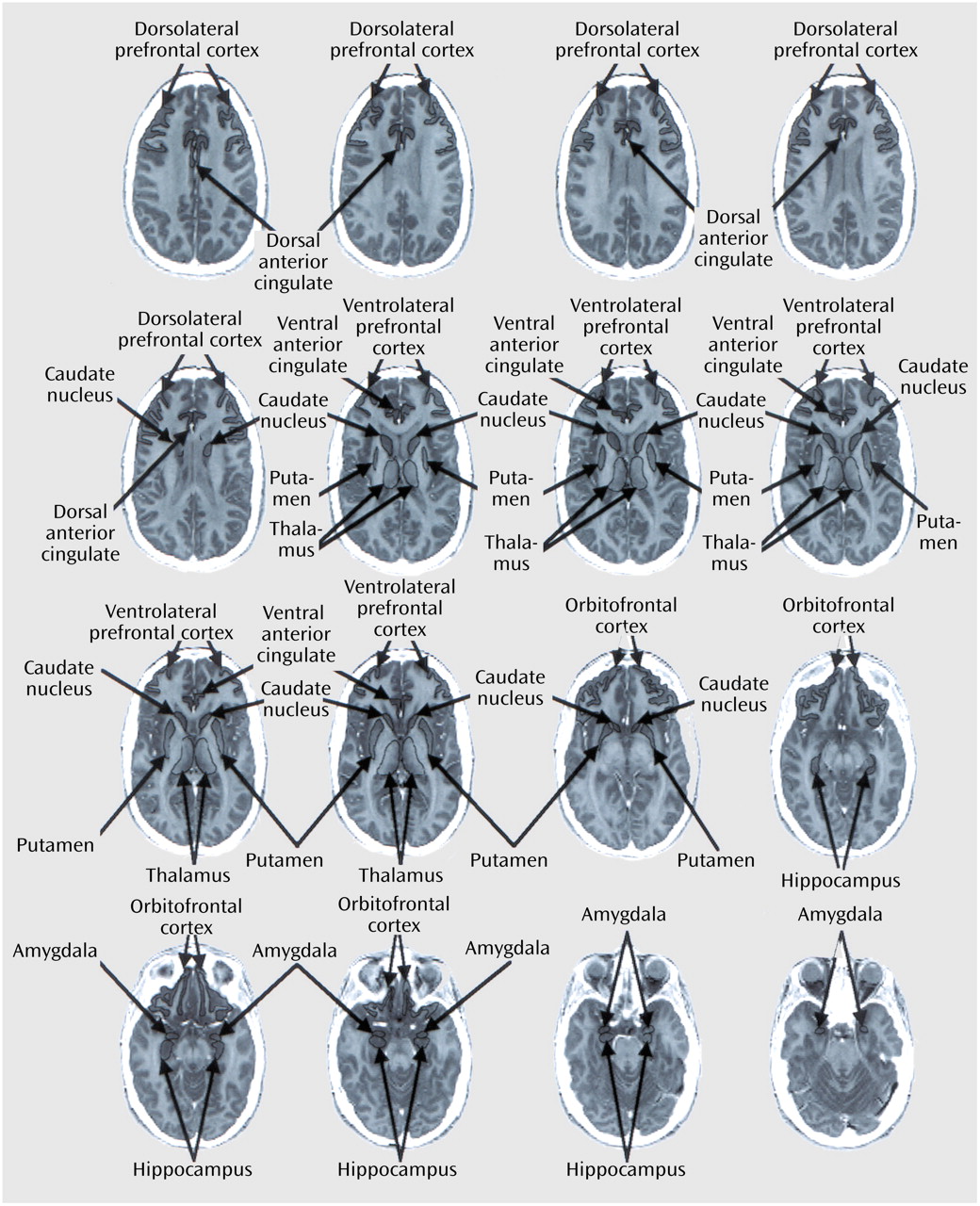
Many methods have been employed to investigate neurobiological predictors of response to antidepressant medications in OCD and major depressive disorder, including neurotransmitter and receptor assays
(3–
8), neuroendocrine challenge tests
(9–
13), neurocognitive testing
(14,
15), auditory evoked potentials
(16), quantitative electroencephalography
(17–
20), and pharmacogenetics
(21–
23). Unfortunately, few of these findings have been replicated, and none have localized specific neuroanatomical substrates for treatment response. Neuroimaging studies, however, have identified pretreatment functional neuroanatomical markers of treatment responsiveness, particularly for SRIs.
In major depressive disorder, pretreatment activity in the midline prefrontal cortex and anterior cingulate gyrus has repeatedly been associated with treatment outcome, but with divergent results. Elevated pretreatment metabolism in the rostral anterior cingulate gyrus was found in responders to SRIs, tricyclic antidepressants, and bupropion
(28), and higher pretreatment glucose metabolism in the left gyrus rectus was correlated with better response to sertraline
(29). In contrast, other studies found that lower pretreatment metabolism in the left ventral anterior cingulate gyrus correlated with better antidepressant response to paroxetine in patients with major depressive disorder
(30) and that baseline metabolism in the prefrontal cortex, anterior cingulate gyrus, and paralimbic structures was lower in responders to venlafaxine and bupropion
(31). Limbic structures may also be involved in responsiveness to antidepressant treatment, as patients with treatment-resistant depression showed higher activity in the hippocampus-amygdala area, compared with patients with non-treatment-resistant depression, in one study
(32).
Discussion
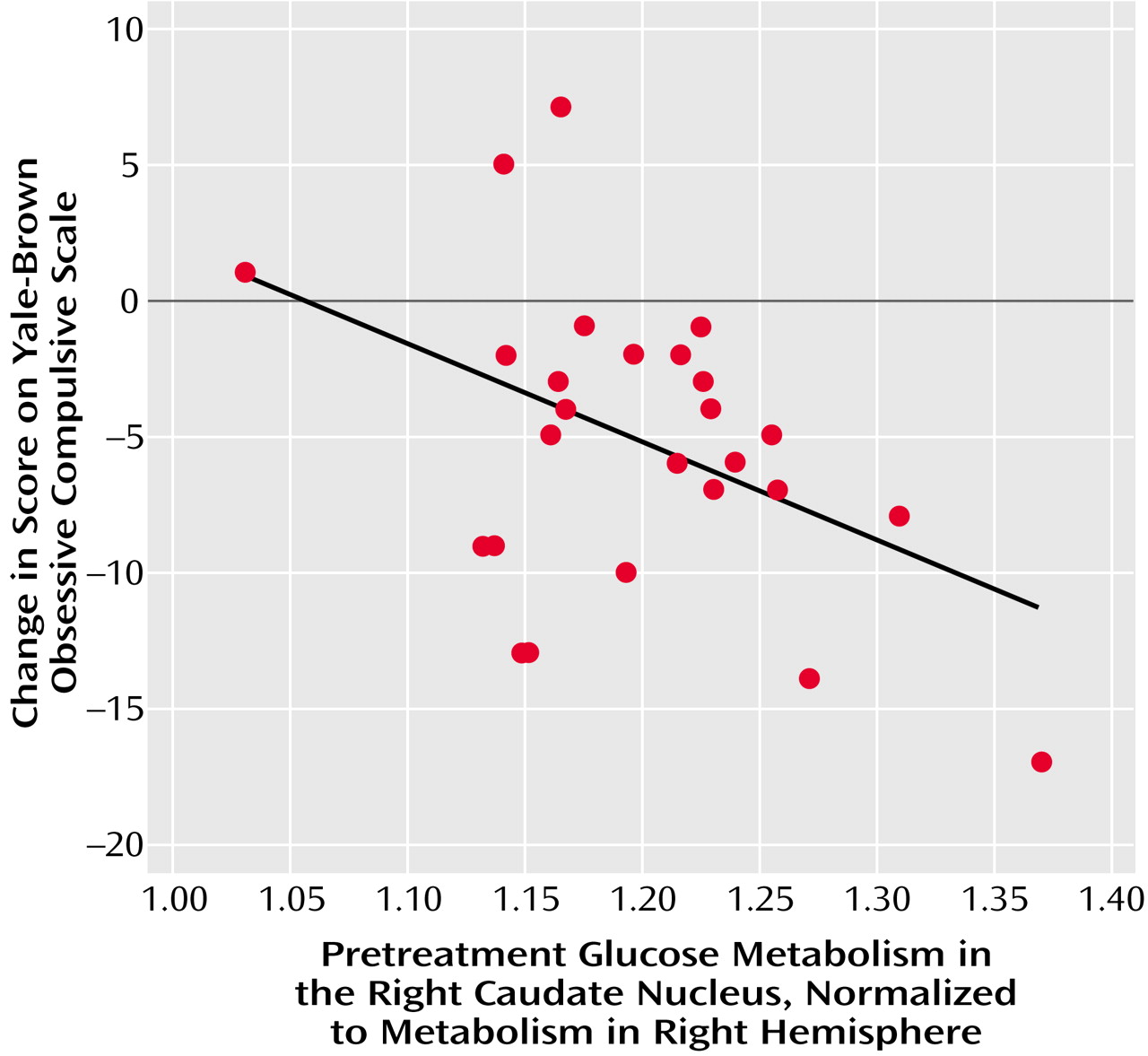
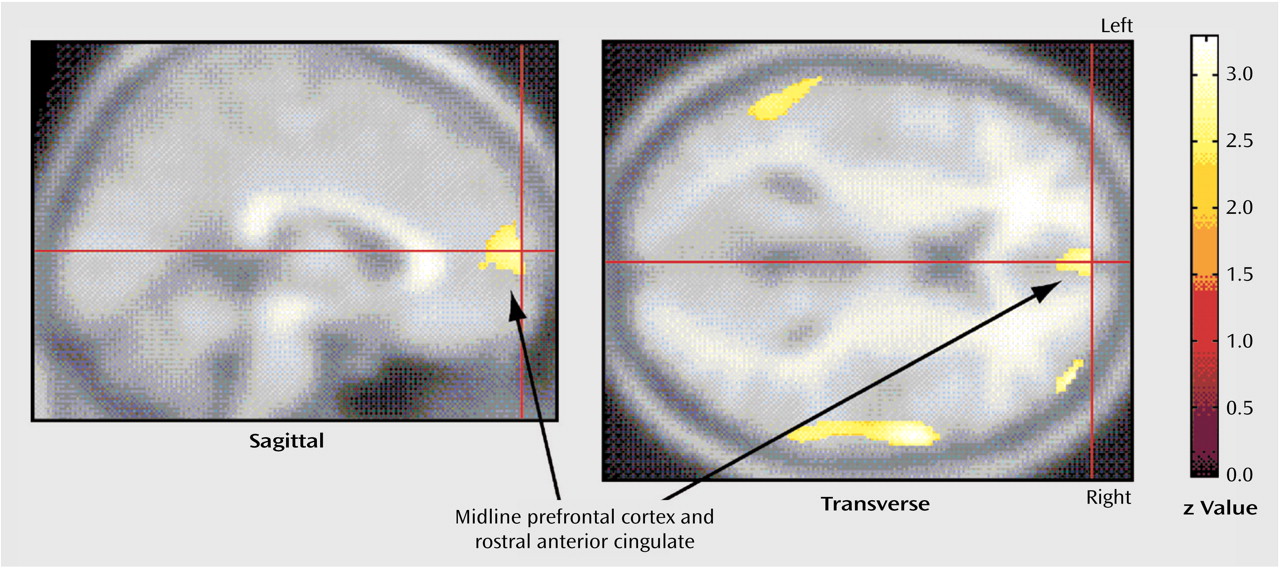
The primary finding of the present study was that different patterns of pretreatment brain activity correlated with the response of different neuropsychiatric syndromes to the same medication, paroxetine. While improvement of OCD symptoms was uniquely associated with higher pretreatment glucose metabolism in the right caudate nucleus, improvement of major depressive disorder symptoms was associated with lower pretreatment glucose metabolism in the amygdala and higher metabolism in the medial prefrontal cortex and rostral anterior cingulate gyrus. These findings suggest that, although both OCD and major depressive disorder respond well to the same SRI medications, the two syndromes have quite different neurobiological substrates for response.
In this study, higher pretreatment metabolism in the right caudate correlated with the degree of improvement of OCD symptoms with paroxetine treatment. Higher glucose metabolism in the right caudate appears to reflect greater release of glutamate
(27,
59), and our findings closely parallel the association between higher pretreatment glutamate concentration in the right caudate and response to paroxetine found by Rosenberg et al.
(27). The right caudate is one of the brain regions most consistently associated with OCD and its response to treatment. Numerous prior neuroimaging studies of OCD patients have found higher levels of activity in the right caudate at baseline (see reference
60 for review) that decrease in responders to a variety of treatments
(61,
62) and increase with symptom provocation
(63). Our group previously reported that treatment-responsive OCD patients were characterized by significant, pathological correlations between glucose metabolic rates in the right caudate, orbitofrontal cortex, anterior cingulate gyrus, and thalamus that were not present in OCD nonresponders, comparison subjects, or patients with major depressive disorder
(61,
62). Thus, higher levels of activity in the right caudate and abnormally strong functional interconnectivity between this structure and other structures along limbic and paralimbic frontal-subcortical circuits
(64) may represent the neural substrate for treatment-responsive OCD. We and others have postulated that overactivity along these circuits may be due to an imbalance of direct versus indirect striatopallidal pathway tone
(60,
65).
We did not fully replicate previous findings of a significant association between pretreatment orbitofrontal cortex metabolism and better response of OCD symptoms to treatment
(24–
26). In the 27 patients with OCD alone in this study, lower normalized pretreatment activity in the orbitofrontal cortex was weakly associated with a decline in Yale-Brown Obsessive Compulsive Scale scores (partial r=0.30, df=21, p=0.16) and an increase in GAS scores (partial r=–0.35, df=21, p=0.10), but these correlations did not reach statistical significance. In all 44 patients with OCD (with and without comorbid major depressive disorder), normalized pretreatment activity in the orbitofrontal cortex was associated with an increase in GAS scores (partial r=–0.32, df=37, p=0.04) but not with a change in Yale-Brown Obsessive Compulsive Scale scores. Methodological differences could account for the discrepancy in results between the present study and prior studies. Most previous studies either analyzed regions of interest drawn on PET scans or did whole-brain searches by using statistical parametric mapping, rather than employing structural MRI-based localization of cerebral cortical regions of interest, as in this study. Symptomatic differences between the subject pools of different studies and differences in the medications used might also contribute to the differences in findings
(60).
Our results add to evidence indicating that higher pretreatment activity in midline prefrontal cortex and anterior cingulate gyrus is a reliable marker of responsiveness to antidepressant treatment. In this study, pretreatment glucose metabolism in a region encompassing the most rostral, affective subdivision of anterior cingulate gyrus
(66) and the medial prefrontal cortex immediately anterior to it was strongly correlated with the degree of improvement of depressive symptoms after paroxetine treatment. A higher level of activity in this same brain region has been associated with eventual response to antidepressant treatment in at least six separate studies that used different treatment and imaging modalities
(28,
29,
67–70). The medial prefrontal cortex and the rostral anterior cingulate gyrus are thought to mediate affective responses
(71) and cognitive/emotional interactions
(72). These regions have heavy connections with the amygdala, hypothalamus, and autonomic brainstem nuclei, as well as with the dorsal and subgenual anterior cingulate gyrus
(71). Mayberg et al.
(28) speculated that adaptive hypermetabolic changes in the rostral anterior cingulate gyrus might be required to facilitate response to treatment. Dopaminergic input to the anterior cingulate gyrus and the medial prefrontal cortex may also modulate response to treatment. Anterior cingulate gyrus metabolism is closely related to dopamine D
2 receptor binding
(73), which was found to increase after SRI treatment of major depressive disorder in responders but not in nonresponders
(74).
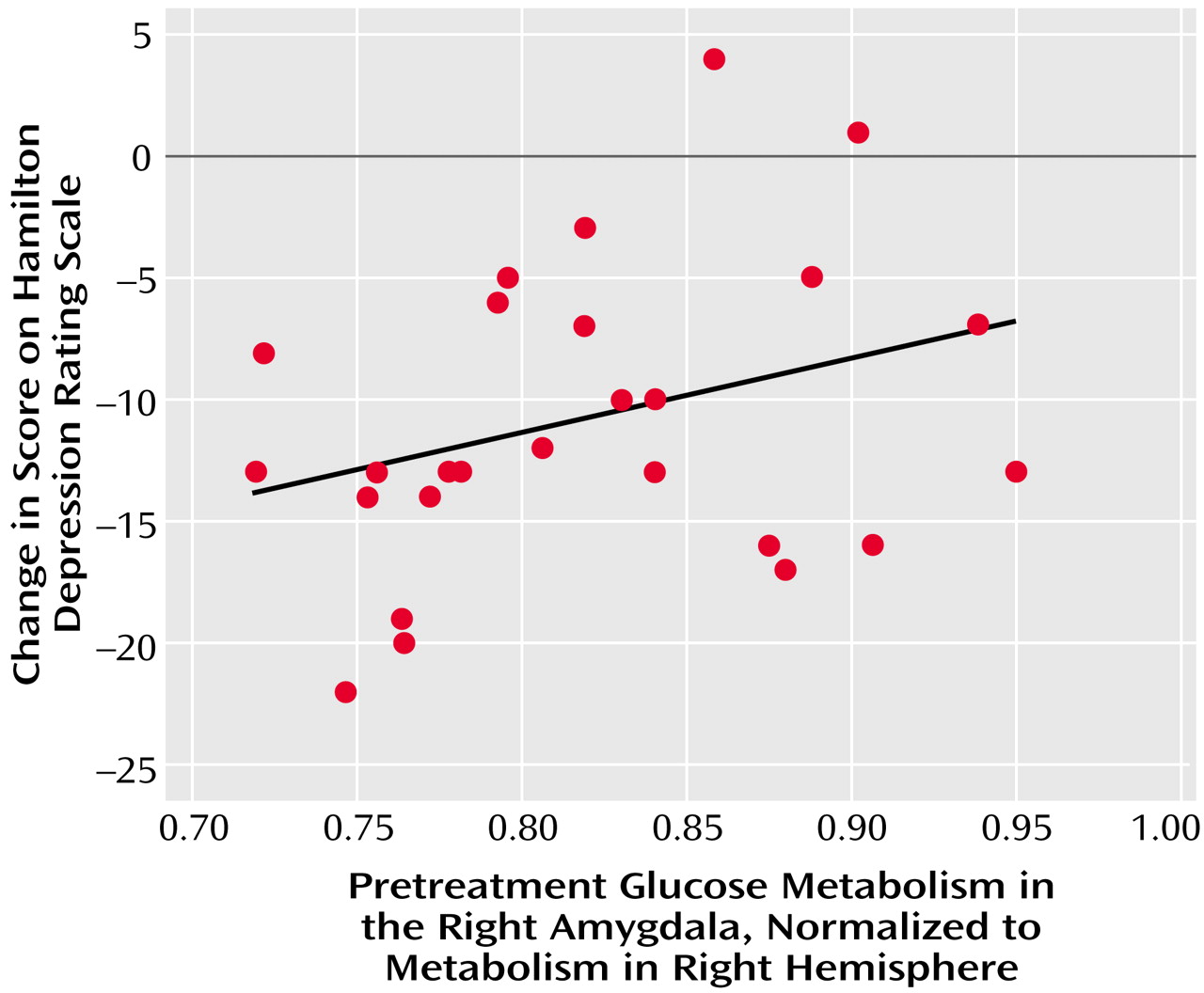
Lower pretreatment activity in the right amygdala was strongly associated with improvement in depressive symptoms in this study, both in patients with major depressive disorder and those with OCD. A large body of evidence points to the central role of the amygdala in the perception of threat, fear, reward conditioning, the assignment of emotional significance to stimuli, and the initiation of emotional responses to those stimuli (see references
75 and
76 for review), as well as in mediating the symptoms of major depressive disorder
(77). The right amygdala shows an ability to habituate rapidly to emotionally valenced stimuli
(78–
80). This decrease of activity in the amygdala is thought to be due to greater inhibition of this structure by the medial prefrontal cortex, an adaptive process by which emotional responses are attenuated and modulated
(81). Thus, lower activity in the right amygdala and higher activity in the medial prefrontal cortex at baseline might represent an ability to inhibit or habituate to emotional responses, allowing an individual to recover eventually from negative emotional states. This may be a neural substrate required for successful response of depressive symptoms to treatment.
Moreover, it has been suggested that abnormally high levels of activity in the amygdala could drive the propensity to relapse or recurrence of depression
(82,
83). During depressive episodes, metabolism in the amygdala correlates positively with both negative affect and plasma cortisol levels
(84), so higher metabolism in the amygdala may indicate a lesser ability to normalize glucocorticoid function during antidepressant treatment, resulting in less improvement in depressive symptoms. Previous findings of higher levels of activity in the amygdala
(32) and abnormal hypothalamic-pituitary-adrenal axis activity
(85) in patients with treatment-resistant depression support this hypothesis. Higher levels of activity in the amygdala may also be related to lower levels of serotonin (5-HT) 1A receptor binding
(86), which in turn could result in poorer response to antidepressants.
Some of the relationships found between pretreatment regional activity and response of major depressive disorder versus OCD symptoms to treatment appear to hold true across diagnostic boundaries. Significant correlations between change in Hamilton depression scale scores and pretreatment glucose metabolism in the amygdala and midline prefrontal cortex were present not only in patients with major depressive disorder alone but also in patients with comorbid OCD and major depressive disorder. Similarly, significant correlations between normalized pretreatment activity in the right caudate and change in Yale-Brown Obsessive Compulsive Scale scores were found in both depressed and nondepressed subjects with OCD. Thus, higher activity in the midline prefrontal cortex/anterior cingulate gyrus and lower activity in the right amygdala appear to reflect the responsiveness to treatment of brain systems mediating depressive symptoms, regardless of primary diagnosis. In contrast, higher levels of baseline activity in the right caudate may reflect the responsiveness of brain systems mediating OCD symptoms to SRI treatment, regardless of the presence or absence of comorbid mood disorders.
Both region of interest and statistical parametric mapping analyses revealed a significant association between lower pretreatment thalamic metabolism and greater improvement in depressive symptoms in patients with OCD (with and without comorbid major depressive disorder) but not in patients with major depressive disorder alone, suggesting that pretreatment thalamic activity may influence treatment response in OCD but not in major depressive disorder. This result is consistent with our previous finding that depression severity was associated with lower thalamic metabolism in patients with OCD, but with higher thalamic metabolism in patients with major depressive disorder alone
(38). Thus, both the severity and responsiveness to treatment of depressive symptoms appear to be mediated differently in patients with OCD than in patients with major depressive disorder.
The present study had several methodological limitations. Because prediction of response was not this study’s original objective, the overall study design did not lend itself to a definitive determination of cerebral metabolic predictors of response, only associations between pretreatment metabolism and treatment response. Therefore, these correlations were secondary analyses, and their results must be interpreted with caution. A more definitive determination of cerebral metabolic predictors of treatment response would require a randomized, prospective, placebo-controlled design with a larger number of subjects. Correction for multiple comparisons was used for the statistical parametric mapping analyses but not for the MRI-based region of interest analyses because the regions of interest were selected on the basis of a priori hypotheses. The large number of correlations performed could potentially have led to type I error in the MRI-based region of interest analyses. However, the statistical parametric mapping and region of interest methods yielded several results in common, including correlations between improvement in Hamilton depression scale scores and lower pretreatment activity in both the amygdala and thalamus. Discrepancies between the results of the region of interest analyses and statistical parametric mapping analyses could have been due to several factors, including statistical parametric mapping’s difficulty in correcting for interindividual variations in neuroanatomy (e.g., in the size, shape, and position of the caudate nucleus), the significance and extent thresholds used for the statistical parametric mapping analyses, and the size and boundaries of the regions of interest.
Another potential limitation was that two different PET tomographs were used in the study. Because scanner type was found to have a significant effect on glucose metabolism in several regions of interest, all data analyses had scanner type as a covariate, which limited the power to detect significant correlations. We analyzed only normalized metabolic rates because the absolute and global metabolic rates generated by the PET methods we used were not felt to be reliable
(33,
34). Normalized and absolute rates have shown different results in prior studies
(60) and may have given different results for this study. In addition, the content of subjects’ thoughts during the FDG uptake phase was not monitored, so the extent to which pretreatment cerebral metabolic patterns reflected specific thoughts and emotions occurring during the scan could not be determined. Because of these limitations, caution is required in drawing firm conclusions from these results.
However, this study also had several strengths that afford confidence in its findings. To our knowledge, this is the largest study of its kind reported, with the largest numbers of OCD and major depressive disorder subjects ever assessed before and after standardized treatment, and it is the first to compare predictors of response to the same treatment across different disorders. All subjects were free from psychotropic medications for at least 4 weeks (and at least 5 weeks for fluoxetine), reducing the risk of medication effects on baseline cerebral metabolism. No other medication but paroxetine was allowed during the study, eliminating confounds from polypharmacy and allowing us to directly compare neurobiological substrates for response to the same treatment in OCD versus major depressive disorder. MRI-based localization of regions of interest was used for the calculation of regional metabolic rates. Statistical parametric mapping and region of interest analyses were compared and produced similar results.
In conclusion, our results support the role of functional neuroimaging in identifying the differential neural substrates of treatment responsiveness in OCD and major depressive disorder. Prediction of treatment response on the basis of pretreatment brain activity could become increasingly important in the future for understanding the mechanisms of treatment response and developing better interventions for patients with treatment-resistant disorders.