Treatments for bipolar disorder include anticonvulsants and atypical antipsychotic agents with antimanic and possible mood-stabilizing effects
(1). In the United States, the anticonvulsant divalproex sodium is widely used because of its proven antimanic activity
(2,
3) and evidence of long-term protective effects against recurrences of bipolar disorder
(4,
5). Its effects are dependent on dose and serum concentration
(6), and rapid antimanic effects are achieved with loading doses of 20 mg/kg per day
(7). Divalproex is generally well tolerated, but sedation, nausea, weight gain, elevated liver enzymes, and thrombocytopenia occasionally lead to discontinuation. Adverse effects and use of multiple daily doses can compromise adherence.
A new extended-release form of divalproex sodium appropriate for once-daily dosing was recently approved by the U.S. Food and Drug Administration for migraine and is approved for epilepsy in Canada. This new form of the medication may offer advantages over standard divalproex, including more stable blood concentrations and simplified, once-daily dosing
(8). An unblinded study found no differences in efficacy or adverse effects between extended-release and standard preparations in patients with epilepsy
(9).
Bioavailability of extended-release divalproex sodium is approximately 85% that of standard divalproex sodium
(10). At steady-state, the extended-release preparation yielded about 15% less fluctuation of average serum concentrations of valproic acid than standard divalproex
(10). A pharmacokinetic study with healthy volunteers found that about 16% greater daily doses of the extended-release preparation were required for serum concentrations equivalent to those of the standard agent
(8). Serum sampling is preferred at 24 hours after the last dose of the extended-release preparation, compared with 12 hours with the standard agent
(8,
10).
The new formulation may be useful in the treatment of bipolar disorder and related conditions. To our knowledge, this is the first report on such use in outpatients formerly maintained on stable doses of standard divalproex.
Method
This open-label, 6-week, pilot study at the bipolar disorder clinic of McLean Hospital involved 12 currently euthymic, clinically stable outpatients (eight women, four men). The patients’ mean age was 45.7 years (SD=11.2). Their DSM-IV diagnoses were bipolar I or II disorder (eight patients, three with rapid cycling) or schizoaffective disorder, bipolar type (four patients). The 12 patients had a mean of 5.7 manic (SD=6.4), 3.7 depressive (SD=4.3), and 2.5 mixed (SD=3.1) previous episodes and a mean of 8.3 hospitalizations (SD=11.2) over a mean of 5.6 years (SD=5.0). The patients were being maintained at clinically individualized, twice-daily divided doses of regular divalproex sodium; their serum valproic acid concentrations were 50–120 μg/ml at 12 hours after the final dose, and they had received stable doses for 4 weeks or longer. Subjects received a mean of 1.9 additional psychotropic medications (SD=1.2) (including a second mood stabilizer in six), all of which were held constant throughout the study.
Exclusion criteria were age less than 18 or more than 65 years, unstable medical illness, previous thrombocytopenia or current hepatic impairment, pregnancy or lactation, current substance abuse disorder, and lamotrigine use. Subjects provided written, informed consent following review and approval of the study protocol by the McLean institutional review board.
In changing from standard to extended-release divalproex, doses were rounded to the nearest 500 mg/day because extended-release divalproex sodium was available only in 500-mg tablets. Regular divalproex sodium was given twice daily, and extended-release divalproex sodium was given once daily at bedtime. A commercial laboratory assayed serum valproic acid concentration by fluorescence polarization immunoassay
(11) of blood samples collected at baseline, day 7, week 6, and 1 week after any dose adjustment (occurring in five patients). Serum was sampled at 10–14 hours after the second daily dose of standard divalproex and 22–26 hours after extended-release divalproex
(8–
10).
Clinical status was evaluated at baseline and weekly with the Young Mania Rating Scale
(12), 17-item Hamilton Depression Rating Scale, Clinical Global Impression (CGI) of severity and improvement, Global Assessment of Functioning Scale (DSM-IV, p. 32), and the 17-item Brief Psychiatric Rating Scale (BPRS), as well as the Udvalg for Kliniske Undersøgelser (UKU) Side Effect Rating Scale
(13) for adverse effects. Laboratory tests (baseline versus endpoint) were a 13-item clinical chemistry panel (including aspartate aminotransferase, alkaline phosphatase, and LDH) and complete blood cell count with differential and platelet count. Baseline-endpoint comparisons used paired t tests; drug level versus dose relationships were assessed by Spearman rank correlation (r
s). Commercial programs—Stata (College Station, Tex., Stata Corp.) and Statview 5 (Cary, N.C., SAS Institute)—were used.
Results
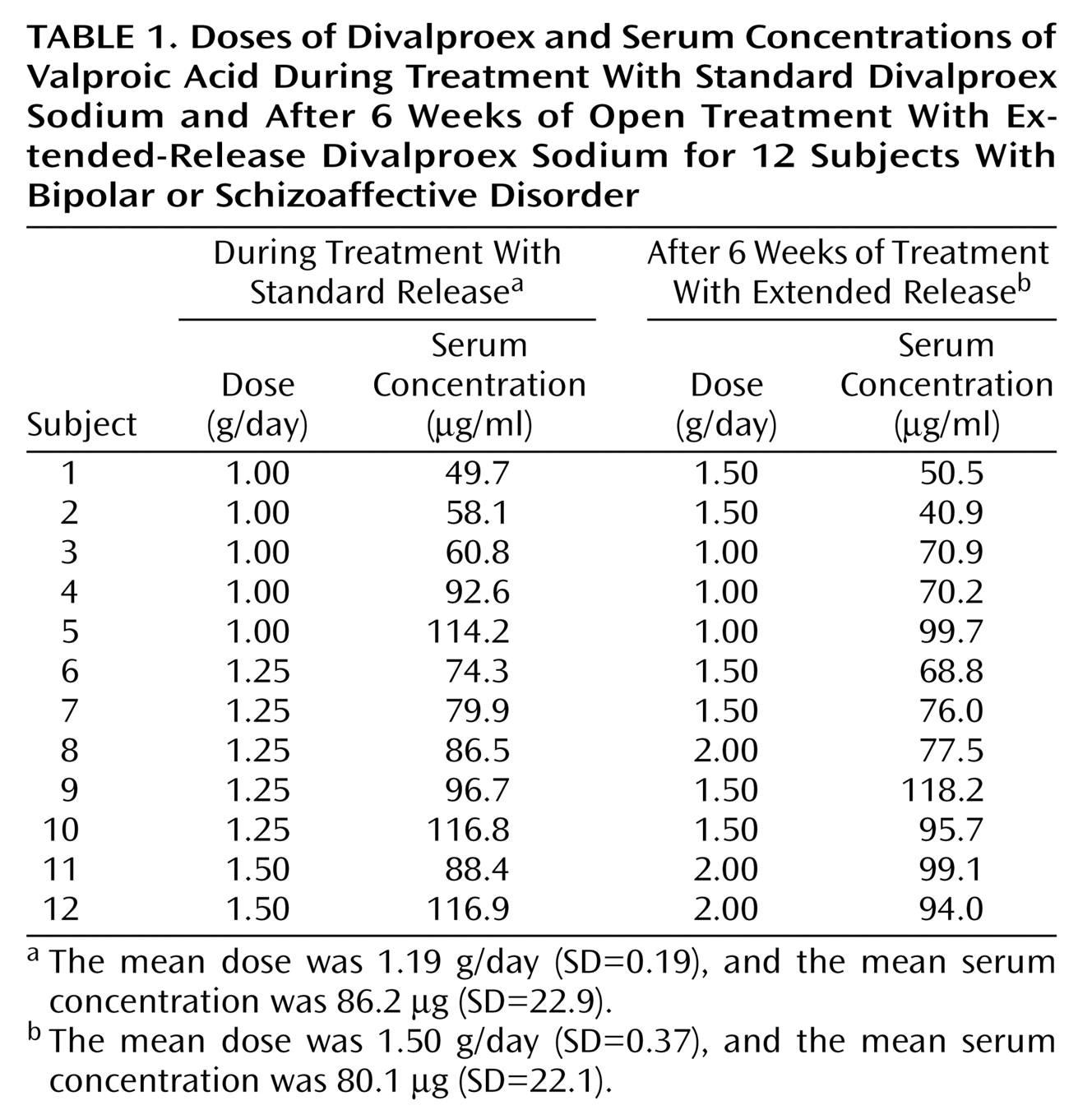
With efforts to maintain stable serum concentrations of valproic acid, individualized daily doses of standard versus extended-release divalproex averaged 1.19 g (SD=0.19) versus 1.50 g (SD=0.37): the extended-release preparation was 20.7% higher (paired t=4.49, df=11, p=0.0009) (
Table 1). Serum valproic acid concentrations were slightly but significantly lower (5.5%) at week 1 (mean=70.6 μg/ml, SD=23.1) than at baseline (mean=86.2 μg/ml, SD=22.9) (paired t=2.68, df=11, p=0.02). After we adjusted daily doses of extended-release divalproex, however, final (mean=80.1 μg/ml, SD=22.1) and baseline (mean=86.2 μg/ml, SD=22.9) serum drug levels were very similar (paired t=1.46, df=1, p=0.17).
Since doses were clinically individualized, a close relationship between daily drug dose and blood concentration was not expected. In fact, according to observations available on both variables at weeks 1 and 6, there was a weakly positive correlation between 24-hour extended-release dose and valproic serum concentration (rs=0.28). Because there was nonindependence within subjects in observations at weeks 1 and 6, this correlation was assessed by random effects regression modeling that took clustering within subjects into account (z=2.39, df=11, p=0.02).
Ratings indicated no significant differences between initial and later clinical status. Mean baseline versus final scale scores were as follows: Young Mania Rating Scale: 3.00 (SD=3.86) versus 3.42 (SD=2.53); Hamilton depression scale: 11.2 (SD=9.3) versus 7.67 (SD=6.97); CGI of severity: 2.58 (SD=0.79) versus 2.75 (SD=0.65); Global Assessment of Functioning Scale: 68.3 (SD=6.2) versus 69.2 (SD=6.0); and BPRS: 39.8 (SD=10.2) versus 37.8 (SD=7.82). Adverse effect summary ratings showed unexpected but significant apparent improvement at week 1 (baseline versus week 1 UKU Side Effect Rating Scale total score: mean=11.2, SD=7.5, versus mean=7.58, SD=3.5) (paired t=2.68, df=11, p=0.02) but not at later weeks or endpoint. Commonly reported adverse events were the same at baseline and endpoint: impaired concentration, fatigue, depression, and decreased salivation. The only significant change was an increase in polyuria-polydipsia (z=2.42, df=11, p=0.03). All subjects elected to continue the extended-release product after completing the trial.
Discussion
In this brief, preliminary report, we found that 12 psychiatric patients were readily switched from standard to extended-release divalproex without evidence of loss of clinical benefit or increased risk of adverse effects within 6 weeks and that all elected to continue with this preparation. Approximately 21% higher daily doses of the extended-release formulation were required to provide equivalent serum concentrations of valproic acid. Simplified, once-daily dosing with the new product should promote medication adherence with potential gains in long-term benefits. These promising findings encourage randomized, controlled evaluations of the extended-release preparation of divalproex in patients with bipolar disorder.