Huntington’s Disease: Phenomenological Diversity of a Neuropsychiatric Condition That Challenges Traditional Concepts in Neurology and Psychiatry
Case History
The present report describes the case of Mr. A, a 49-year-old man with rapidly increasing physical and mental impairments who had been diagnosed with Huntington’s disease 1 year before admission to our department. Analysis of the patient’s medical history revealed a long history of various neuropsychiatric disturbances, diagnoses, and treatment approaches preceding the differential diagnosis and genetic verification of the disease (Table 1).
Personal and Social Background
Mr. A was born in 1950 after an uncomplicated pregnancy, labor, and delivery. His natural father had abandoned the patient’s family and three other siblings when he had been only 3 years old. According to his mother, he was an anxious and bed-wetting child who was often the target of his stepfather’s violent outbursts. At the age of 12, he exhibited recurrent cataplectic attacks with a sudden loss of muscle tone and immobility accompanied by unaltered consciousness. Although he was actually reported to be an intelligent and successful student, the frequency of the attacks disabled the boy so much that he had to repeat a school year. The results of a standard neurological examination yielded no obvious irregularities, so the symptoms were attributed to an “unknown psychological causation.” Along with his remarkable family background, this diagnosis may have misguided the subsequent appraisals of his therapists. At age 20, after successful completion of secondary school and training as an insurance agent, Mr. A got married. According to his wife, he exhibited dependent and jealous behavior right from the beginning of their relationship. Apart from this, premorbid social and occupational functioning was always unobtrusive. During 1990, our patient experienced various phobic symptoms of increasing severity, leading to prolonged sick leave and inpatient treatment after acute exacerbation of the symptoms and separation from his spouse. As a result of the unknown fate of the patient’s biological father, his family history remained incomplete. None of Mr. A’s available relatives was afflicted with a neuropsychiatric disorder.
Neuropsychiatric Development
At the time of his first admission in 1991, Mr. A showed signs of intense fear, including tachycardia, hyperhydrosis, and extreme stuttering, which prevented any reasonable exploration before the application of benzodiazepines. He reported unspecific “worryness” and recurrent states of massive anxiety, which were generally but not exclusively situation-bound (e.g., in darkness, crowded rooms, or while driving a car). No clear psychotic symptoms were seen at that time.
Initial Diagnosis: Neurotic Anxiety
The clinical examination of Mr. A’s neurological and electroencephalographic status yielded no pathological findings, a computerized tomography (CT) scan equally failed to reveal any striking irregularities. In addition to an average IQ level, his neuropsychological screening revealed varied results regarding Mr. A’s attentional functions. This inhomogeneous test profile was obviously caused by inferior test performance in all speed-dependent subscales (>1.5 SDs below mean performance). Unfortunately, this pathognomic indicator was not traced further. Instead, the therapist’s attention was attracted by the “obvious childhood origin of the neurotic symptomatology,” and a neurotic anxiety disorder was subsequently diagnosed (DSM-III-R, code 300.0). The therapeutic regime consisted of a β-blocking intervention with propranolol hydrochloride and referral for outpatient behavioral therapy. In the absence of clear psychotic symptoms, further progression of the clinical picture was characterized by various psychiatric symptoms exacerbating as a result of environmental stressors, a gradual increase of subtle cognitive and psychomotor constraints, and growing dependence concerning daily demands.
Second Diagnosis: Endogenous Depression
Approximately 4 weeks later, Mr. A had to be readmitted in a desolate state after he had started to sleep on park benches after a severe dispute with his mother. Retrospectively, Mr. A reported a broad array of psychiatric symptoms, including feelings of insufficiency, helplessness, and pronounced attentional deficits (Table 1). Intense psychomotor restlessness prevented him from sitting down during meals or conversation. For understandable reasons, Mr. A’s thoughts were narrowed to worries concerning his medical problems and the painful separation efforts of his wife. Unfortunately, the professional’s view of that time remained overly focused on Mr. A’s “extensive neurotic family background,” and his worries were interpreted as “pronounced hypochondriac ideas.” At this time, his complaints were classified as endogenous depression (DSM-III-R, code 296.2) and treated thymoleptically with amitriptyline.
Third Diagnosis: Ganser Syndrome
In December 1994, Mr. A was admitted to the hospital with a presumed psychotic disorder of organic origin. As a result of the continuous quarrel with his relatives, he had become homeless and repeatedly spent the night in his car. On admission he was disoriented, exhibited a partially clouded consciousness, psychomotor slowing, and a seesaw gait disturbance. During the clinical interview, the psychiatrist adopted previous psychodynamic explanations and focused on the pronounced approximate answers of the patient, especially when his marital relationship was addressed. More precisely, the lack of insight concerning separation efforts by his wife left the impression of an intentional aggravation of his complaints in terms of a maladjusted defense style. This interpretation was supported by the psychological disclosure of a tendency to pattern his behavior according to its social desirability (MMPI). Subsequently, a dissociative disorder specified as “Ganser syndrome” (DSM-III-R, code 300.15) was diagnosed, and the basic necessity for a long-term psychotherapeutic confrontation with his adaptive difficulties was advised.
Fourth Diagnosis: Schizophrenia
Four weeks later, Mr. A was hospitalized in yet another state mental institution with pronounced affective depletion, substantial loss of impetus, and psychomotor slowing interpreted as residual negative symptoms. For the next 2 years, he remained hospitalized with a diagnosis of schizophrenia, the fluctuating clinical picture scarcely stabilized with clozapine treatment (DSM-III-R, code 295.2; DSM-IV, code 295.6). His complaints consisted of diffuse anxiety symptoms, reduced attentional capacities and mental flexibility, as well as increasing hypokinesia and rigidity. Positive psychotic symptoms were present to a lesser extent and consisted of intermittent stages of pronounced distrust and formal thought disorder. In 1996, the patient’s status showed further deterioration, since he had now also begun to neglect his personal hygiene, needed support in managing even trivial demands, and developed dysarthric speech.
Final Diagnosis: Huntington’s Disease
One year later, the clinical picture also included akinesia, ataxia, and urinary incontinence. Even then, his results on conventional neuropsychological tests focused on the assessment of dementia (cognitive section of the Cambridge Examination for Mental Disorders of the Elderly, the Mini-Mental Status Examination [MMSE], the Structured Interview for the Diagnosis of Dementia) and degenerative brain processes were not clearly in the pathological range (Cambridge Examination for Mental Disorders of the Elderly, total score=87; MMSE, total score=26). Genetic testing for Huntington’s disease was not considered until clear choreatic hyperkinesias emerged 7 years after his first psychiatric referral. Despite the patient’s widespread mental and physical disability, standard neurological, neuroradiological, and neuropsychological procedures still provided no reliable evidence for a primary neurodegenerative disease. The origin of the genetic defect was not definitely determinable from the available evidence. Because of the early disappearance of the patient’s father, a de novo expansion of the unaffected parents’ CAG was just as plausible as a paternal transmission of the CAG expansion.
Neuropsychiatric Profile at Current Admission
Mr. A’s last admission to our institute had been initiated by the local police. Now a resident in a rehabilitative institution for psychiatric patients, he was found in a streetcar, mute, petrified, and trembling, unable to give his identity on request. Clinical observations confirmed the initial impression of a further deterioration of his condition. To define his remaining functional capacities more precisely, a detailed psychiatric, neurological, and neuropsychological assessment was carried out.
Psychiatric and Neurological Examination
In addition to previously known cognitive deficits, Mr. A now exhibited a marked gait and balance disturbance, often requiring the use of a wheelchair. Psychopathologically, an overall reduction of apprehension, impetus, affective expression, and attention was concurrent with a tendency for social withdrawal. Although conversation was notably complicated by pronounced dysarthria, he comprehensively reported subjective distress because of his urinary incontinence. Furthermore, a recently emerged swallowing disability had demanded recurrent pulp nourishment and had given rise to persistent asphyxiation fears while he was eating. In contrast to the pronounced slowing of intentional motor functions, choreatic hyperkinesias of the face and trunk were only sporadically observable. Overall, the clinical impression of end-stage Huntington’s disease arose. Therapeutic interventions focused on pharmacological stabilization with clozapine, tiapride, and tetrabenazine.
Neuroimaging
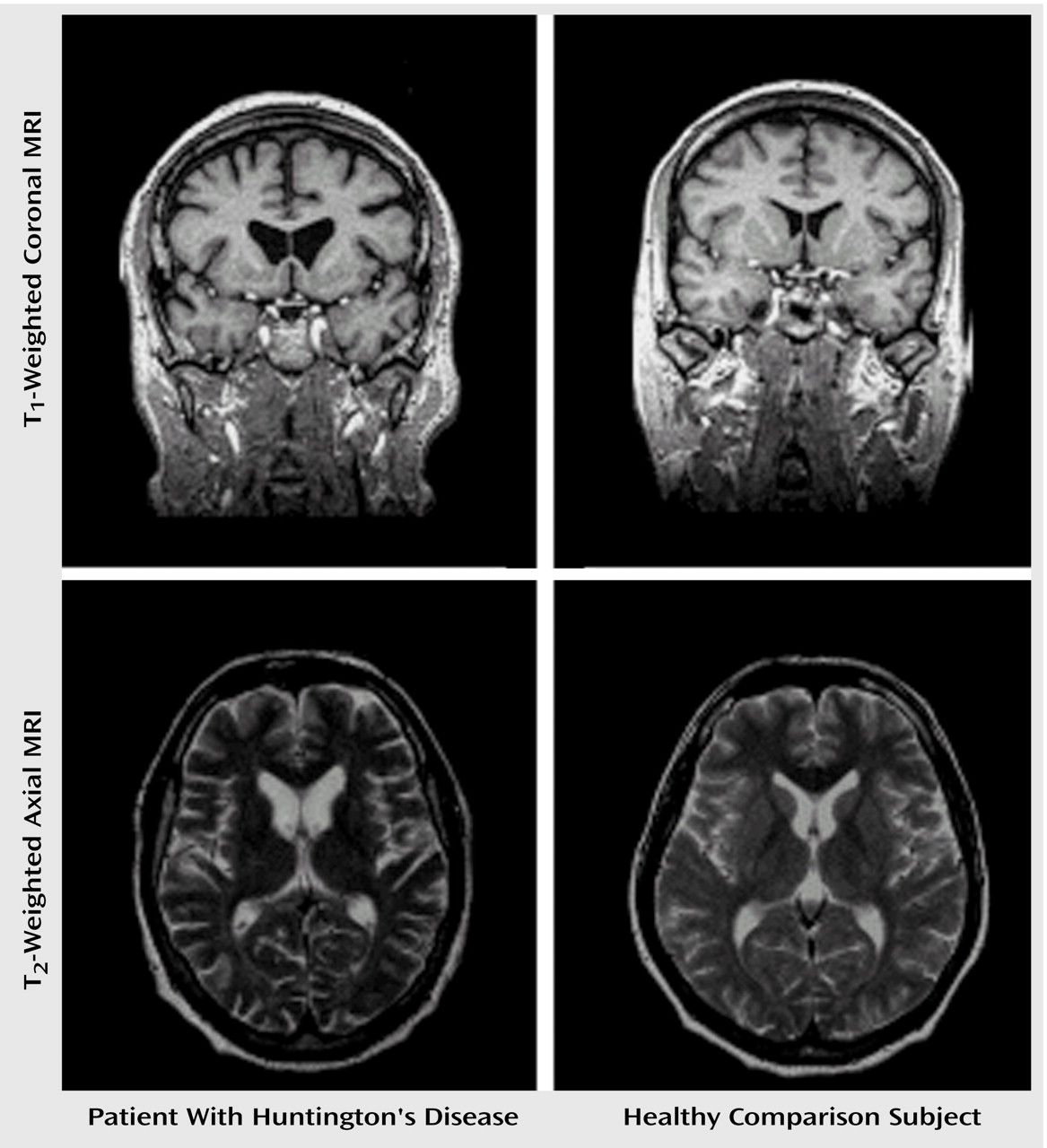
Brain imaging was performed by using standard proton-density-weighted, fluid-attenuated inversion recovery and three-dimensional volumetric magnetic resonance sequences. In contrast to previous CT scans, these morphological pictures verified the characteristic neurodegenerative changes of Huntington’s disease (Figure 1). Because of the considerable atrophy of the head of the caudate nucleus, the frontal horns of the lateral ventricles appeared bilaterally enlarged. An additional target of neuronal loss was localized in the putamen. In contrast to the focal neuronal loss of the striatum, the widespread accentuation of cortical sulci is in line with diffuse cerebral atrophy.
Neuropsychological Findings
Considering the known dementia syndrome secondary to Huntington’s disease, the current cognitive examination focused on the assessment of Mr. A’s mnestic functions. In line with the psychological evaluation 2 years earlier, we performed a structured interview for the diagnosis of dementia of the Alzheimer type, multi-infarct dementia, and dementias of other etiology (Structured Interview for the Diagnosis of Dementia), the Cambridge Examination for Mental Disorders of the Elderly, as well as the MMSE. Problem-solving and executive capabilities were analyzed with the Wisconsin Card Sorting Test (10). Behavioral, functional, and motor abnormalities were estimated by using the Unified Huntington’s Disease Rating Scale (11). Furthermore, a broad range of basic attentional functions were examined by using the computerized Test Battery for the Assessment of Attention (12).In accordance with the clinical impression, the Unified Huntington’s Disease Rating Scale motor examination revealed a moderate movement disability, with pronounced signs of bradykinesia, dysarthria, and coordination deficits (total score=25). The behavioral assessment disclosed mild depressive symptoms (total score=8). In contrast to our initial expectations, the applied dementia ratings again did not indicate the presence of a dementia syndrome. Although a slight decline in nearly all subscales was observable compared to the prior evaluation, the various dementia ratings still did not grasp the obvious cognitive constraints of our patient (Cambridge Examination for Mental Disorders of the Elderly, total score=80; MMSE, total score=24).However, the evaluation of our patient’s basic attentional capabilities (Test Battery for the Assessment of Attention) and executive functions (Wisconsin Card Sorting Test) clearly pointed to the presence of pronounced cognitive and psychomotor deficits. Although Mr. A showed regular reactions to task-contingent external stimuli (phasic alertness value: 76th percentile), marked psychomotor slowing was obvious while we determined his basic reaction speed (his median reaction time was less than the first percentile). His attempts to handle the working memory and response flexibility tasks resulted in a complete performance breakdown, indicating an exceptional dorsolateral-prefrontal processing deficit (omission responses were false alarms of less than the first percentile). The consistently low Wisconsin Card Sorting Test performance scores also revealed apparent difficulties regarding frontally mediated skills, especially cognitive flexibility and abstract reasoning (total categories and perseverations scores: less than the first percentile). In contrast, the arousal-related internal maintenance of attention in the vigilance task was unaffected (omissions and false alarms: 42 responses, 76th percentile). Overall, the neuropsychological profile of Mr. A pointed to a locally constricted frontal processing deficit affecting prefrontal cognitive and motor areas rather than the midtemporal-hippocampal regions usually associated with neurodegenerative memory complaints.
Discussion
Phenomenological Diversity of Initial Huntington’s Disease
Spectrum of Motor Abnormalities
Development of Cognitive Deficits
Early Psychiatric Manifestations
Implications: An Extended Neuropsychiatric Perspective

Footnote
References
Information & Authors
Information
Published In


History
Authors
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
There are no citations for this item
View Options
View options
PDF/ePub
View PDF/ePubGet Access
Login options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).