At present, the only approved pharmacological approach for the symptomatic treatment of Alzheimer’s disease in Japan is the use of cholinesterase inhibitors. Donepezil hydrochloride has been demonstrated to have significant effects in slowing symptomatic progression in 24-week placebo-controlled trials
(1), and some long-term studies have shown that there is no less benefit after 1 year of treatment
(2,
3). One observational study
(4) showed that cholinesterase inhibitor treatment alters the natural history of Alzheimer’s disease, as indicated by the delay in admission to nursing homes. Furthermore, a longitudinal neuroimaging study using single photon emission computed tomography (SPECT) demonstrated that treatment of patients with Alzheimer’s disease with donepezil for 1 year reduced the decline in regional cerebral blood flow (rCBF)
(5), suggesting the preservation of functional brain activity in donepezil-treated patients. Although these studies appear to have demonstrated the efficacy of donepezil in slowing down clinical disease progression, it is not clear whether donepezil treatment slows disease progression in Alzheimer’s disease.
Neuropathologically, Alzheimer’s disease is characterized by the presence of neurofibrillary tangles and senile plaques, impaired synaptic function, and cell loss
(6). Although these histological features cannot be examined noninvasively, the cell loss that accompanies them can be seen in vivo as atrophy with magnetic resonance imaging (MRI). Among the characteristic neuropathological changes in Alzheimer’s disease, the most prominent structural changes at the initial stage occur in the hippocampal formation
(7,
8). MRI-based volumetry of the medial temporal lobe structures has been proposed as a useful tool for the clinical diagnosis of Alzheimer’s disease
(8). Serial MRI studies permit calculation of rates of atrophy over time. It has been proposed that measurement of the rate of atrophy could be used to monitor the effectiveness of antidementia drugs for Alzheimer’s disease
(9,
10). If an anti-Alzheimer drug can slow down the anatomic progression of Alzheimer’s disease pathology, this should be detectable as a decrease in the rate of hippocampal atrophy in treated patients.
The purpose of the present study was to determine whether a cholinesterase inhibitor, donepezil hydrochloride, has a neuroprotective effect on Alzheimer’s disease. Using an MRI-based volumetric technique, we examined the rates of hippocampal atrophy in donepezil-treated Alzheimer’s disease patients and compared the results with those in control patients.
Results
Twelve of 77 patients who were initially enrolled in the donepezil follow-up program were dropped from the study. Seven withdrew their consent, two were institutionalized, and the remaining three withdrew for other reasons. Of the 65 patients who completed the entire procedure of the study, 11 were excluded because the quality of one of their two MRIs was unsuitable for volumetry. Thus, 54 patients remained in the treated group. Two patients did not receive the full, 1-year dosage of donepezil. One patient discontinued donepezil after 3 weeks because of adverse effects, and the other took the drug irregularly (receiving about 60% of the full dose). However, these patients were included in the primary analyses.
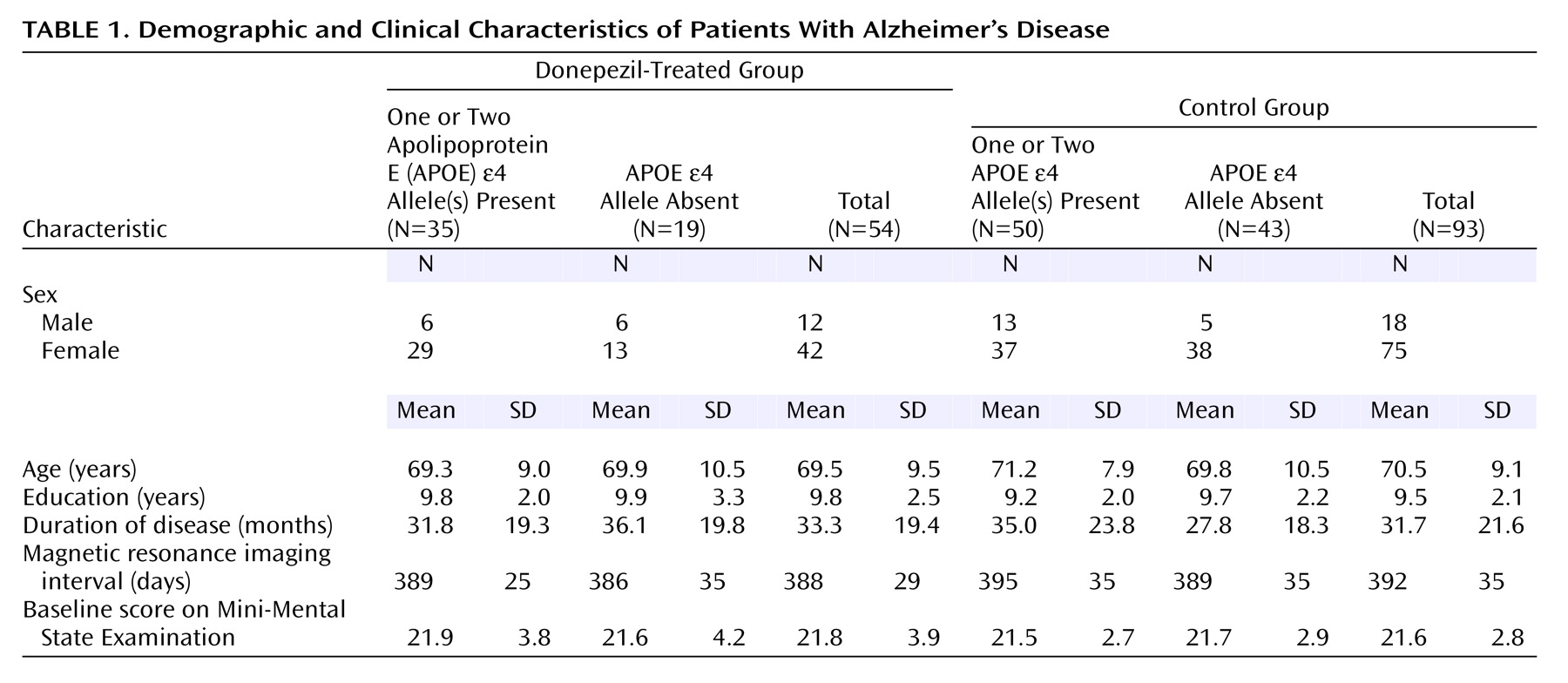
Of the 108 patients who completed the entire procedure of the annual follow-up study, 15 patients were excluded because their MRIs were of poor quality. Thus, 93 patients remained in the control group. The background characteristics of both groups are summarized in
Table 1. The treated and control groups were not significantly different in age, sex, education, disease duration, intervals between the first and second MRI, or baseline MMSE scores.
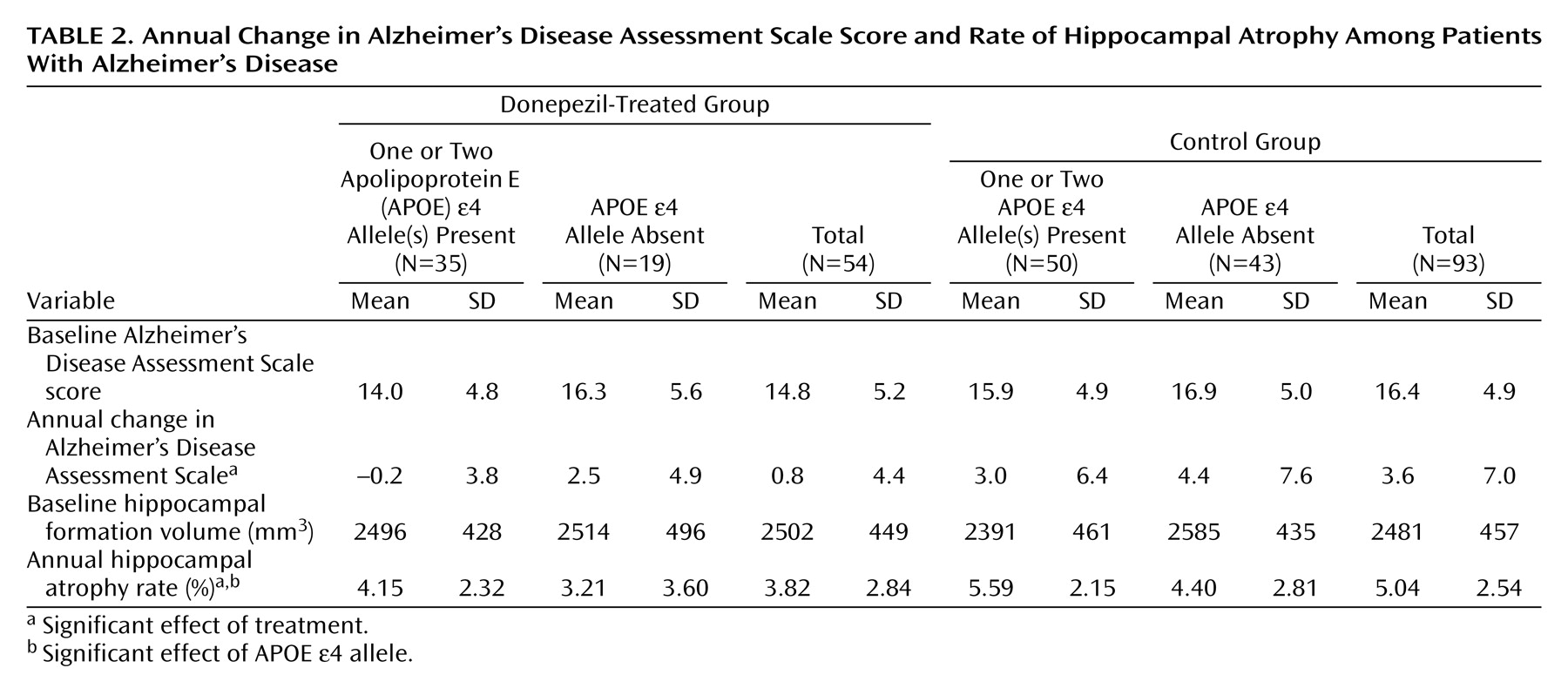
Hippocampal volumes and Alzheimer’s Disease Assessment Scale scores are summarized in
Table 2. The effect of donepezil treatment on the change in Alzheimer’s Disease Assessment Scale scores was significant (F=5.55, df=143, p<0.02), but neither the effect of the APOE e4 allele (F=3.64, df=143, p<0.06) nor the interaction term (F=0.32, df=143, p=0.57) was significant. The effects of donepezil treatment and the APOE e4 allele on the rate of hippocampal atrophy were significant (F=8.19, df=143, p=0.005, and F=5.29, df=143, p<0.03, respectively), but the interaction term was not significant (F=0.08, df=143, p=0.78). In a further analysis, the mean annual rate of hippocampal atrophy in the treated group (mean=3.82%, SD=2.84%) was significantly lower than in the control group (mean=5.04%, SD=2.54%) (t=–2.7, df=145, p=0.008). In a one-way ANCOVA, where age, sex, disease duration, education, MRI interval, APOE genotype, and baseline Alzheimer’s Disease Assessment Scale score were entered into the model as covariates, the effect of donepezil on hippocampal atrophy remained significant (F=10.34, df=138, p=0.002).
Even if the two patients who did not receive the full 1-year dosage of donepezil were excluded from the analyses, the results remained unchanged; donepezil treatment had significant effects on Alzheimer’s Disease Assessment Scale score (F=4.37, df=141, p<0.04) and the rate of hippocampal atrophy (F=6.53, df=141, p<0.02), and the APOE e4 allele had a significant effect on the rate of hippocampal atrophy (F=4.14, df=142, p<0.05). The mean annual rate of hippocampal atrophy in the treated group (mean=3.93%, SD=2.76%) was significantly lower than in the control group (mean=5.04%, SD=2.54%) (t=–2.4, df=143, p<0.02).
Discussion
In the present study, the mean annual decline in the Alzheimer’s Disease Assessment Scale score in the control group (3.6 points) was significantly larger than in the treated group (0.8 points). Thus, these results are consistent with the results of previous long-term studies of donepezil
(2,
3), and suggest that treatment with donepezil for 1 year was associated with a cognitive benefit. Moreover, donepezil seemingly slows the rate of hippocampal atrophy. The decrease in hippocampal volume in the treated patients (3.82%) was about 24% less than in the control patients (5.04%). The present results suggest that donepezil has both symptomatic and disease-progression slowing effects. The mean annual rate of hippocampal atrophy was significantly higher in the patients with the APOE e4 allele than in those without the APOE e4 allele, replicating our previous observation that the APOE e4 allele is specifically related to accelerated hippocampal pathology in Alzheimer’s disease
(9). On the other hand, for the rate of hippocampal atrophy and for the change in Alzheimer’s Disease Assessment Scale, no significant interaction was noted between donepezil treatment and the APOE genotype. In a previous study, the effect of donepezil treatment did not appear to be affected by the APOE genotype
(22). Our data are consistent with this observation.
In recent studies, long-term donepezil treatment has been demonstrated to slow the decline in cognition and functional activities
(2,
3), delay the admission to a nursing home
(4), and reduce the decline in rCBF
(5). However, these studies do not answer the question of whether the beneficial effects of donepezil are due to symptomatic suppression, which leaves the underlying disease process unaltered, or to neuroprotection modifying the disease process. Recently, one randomized, double-blind, placebo-controlled pilot study in patients with Alzheimer’s disease demonstrated that donepezil-treated patients had significantly smaller mean decreases in total hippocampal volumes compared with placebo-treated patients. Although that study had limitations in that it included a relatively small number of patients and the period of drug treatment was short, our data are consistent with the results
(23). Furthermore, two clinical trials, both open-label, placebo-controlled extension studies, have raised the possibility that cholinesterase inhibitors have slowing effects on both symptomatic and disease progression in Alzheimer’s disease patients. In one study
(24), the level of cognitive function in patients who were treated with placebo for 3 months, followed by 12 months of donepezil treatment, was lower than in patients who were treated with donepezil for all 15 months. In the other study
(25), the level of cognitive function of the patients who were treated with placebo for 26 weeks, followed by 26 weeks of rivastigmine treatment, was lower than that in patients who were treated with rivastigmine for all 52 weeks. The inability to catch up in those in whom the treatment with cholinesterase inhibitors was postponed reminds us of a potential disease-modifying effect.
A recent long-term randomized, double-blind trial
(26) showed that no significant benefits were seen with donepezil compared with placebo in the institutionalization and progression of disability. In the study, there were no significant differences in behavioral symptoms, caregiver well-being, and caregiver time costs. On the other hand, statistically significant cognitive and functional effects were maintained over at least 2 years. Because many of the outcomes are influenced by the interaction of complex biological, social, and environmental factors, they might be inappropriate for assessing the neuroprotective effects of donepezil.
To explain the neuroprotective effect of cholinesterase inhibitors, mechanisms based on beta-amyloid metabolism have been postulated. Accumulation of amyloid is one of the earliest changes in Alzheimer’s disease pathology
(27,
28) and may cause neuronal death in the CNS
(29,
30). In vitro studies have demonstrated a link between cholinergic activation and beta-amyloid precursor protein metabolism. Wallace et al.
(31) found evidence that lesions of the cholinergic nucleus basalis of Meynert increased the synthesis of beta-amyloid precursor protein in the cerebral cortex of rats. Wolf et al.
(32), using human CNS neurons, found increased amyloid precursor protein secretion and decreased beta-amyloid protein production with carbachol stimulation of muscarinic receptors
(32). These studies support a beneficial alteration in amyloid processing associated with cholinergic stimulation. Kihara et al.
(33) examined the effects of nicotinic receptor agonists on amyloid beta cytotoxicity in cultured rat cortical neurons and found that nicotinic receptor stimulation may be able to protect neurons from degeneration induced by amyloid beta. Svensson and Nordberg
(34) demonstrated that tacrine and donepezil at clinically relevant concentrations attenuated amyloid beta (25-35)-induced toxicity in rat pheochromocytoma PC12 cells. The neuroprotective effect was blocked by the presence of the nicotinic antagonists mecamylamine and tubocurarine, suggesting an intervention through nicotinic receptors.
Our study has some limitations. First, because it was a comparative study with a historical control group rather than a randomized study, it suffers from several sources of bias. The groups are not comparable because of the selection of subjects who received the intervention (selection bias), the cointerventions and other medical management being received by the two groups were different (performance bias), and the methods of outcome measurement being used in the two groups were different (detection bias). Although there was no intention to select subjects for the control and treatment groups, patients who could not tolerate the initial doses of donepezil were not included in the donepezil follow-up program. This could have been a source of selection bias. However, we are unaware of any evidence that donepezil tolerance predicts disease progression. Because the control group in the present study was not concurrent but historical, there was a generation difference of several years between the groups. Although the generation difference was a possible source of selection and performance bias, the difference in the patients’ generation was not likely to affect the volumetric results, and the cointerventions and other medical management did not change throughout the first and second halves of the study period. Second, it has been recognized that open-label studies are not optimal. The fact that the investigators were not blinded to treatment might increase the donepezil effect. However, neither hippocampal volume nor the Alzheimer’s Disease Assessment Scale score was a subjective outcome measure. Furthermore, in the present study, the Alzheimer’s Disease Assessment Scale was administered by neuropsychologists who were unblinded but not involved in managing the patients, and MRI volumetry was made by an investigator who was blinded to all clinical information. Therefore, it was unlikely that detection bias affected the results of the rate of hippocampal atrophy or the Alzheimer’s Disease Assessment Scale score in favor of the donepezil-treated group. Third, dropouts are a problem for this type of design and a possible source of exclusion bias. Simply ignoring everyone who has withdrawn from a clinical trial may bias the results, usually in favor of the intervention. Therefore, it is standard practice to analyze the results of comparative studies on an intention-to-treat basis. In the present study, two patients who did not take the full 1-year dosage of donepezil because of adverse events or poor compliance were included in the primary analyses.
As a result, the patients’ background characteristics, such as age, sex, education, disease duration, disease severity, and MRI interval, were comparable between the two groups, and even after we controlled for these variables, the effect of donepezil on hippocampal atrophy remained unchanged. In any case, the significant difference of the rate of hippocampal atrophy was likely to be due to the intervention with donepezil. Although our findings should be confirmed by a randomized, controlled long-term trial in patients with Alzheimer’s disease, it would be unethical to conduct such a study to extend the knowledge of donepezil.
The present results suggest that donepezil has not only a symptomatic effect but also a neuroprotective effect. If donepezil does, in fact, influence disease progression, we need to modify our treatment strategies; donepezil is not an optional but rather a mandatory treatment for Alzheimer’s disease and should be started in the prodromal or very early stage of the disease. Mild cognitive impairment is a condition that frequently progresses to Alzheimer’s disease, which requires early diagnostic and therapeutic interventions. Donepezil, through its neuroprotective effect, could possibly inhibit progression from mild cognitive impairment to Alzheimer’s disease. Further studies are needed to determine whether donepezil slows the progression from mild cognitive impairment to Alzheimer’s disease. Hippocampal atrophy could be used as a surrogate marker of disease progression in such studies. Furthermore, the potential neuroprotective mechanism should be refined and exploited to enhance the drug’s effectiveness in treating Alzheimer’s disease. A better understanding of this mechanism may suggest strategies for designing improved drugs.