Since the introduction of selective serotonin reuptake inhibitor (SSRI) antidepressants in the 1980s, there has been controversy over whether they can trigger suicidal thoughts or behavior
(1,
2) . In 1991, the Psychopharmacologic Drugs Advisory Committee of the U.S. Food and Drug Administration (FDA) found no clear evidence of increased suicide risk associated with fluoxetine
(3) . In 2003, however, the U.K. Medicines and Healthcare Products Regulatory Agency concluded that paroxetine, citalopram, and other SSRIs were contraindicated in youths because of an increased risk of treatment-emergent suicidal ideation
(4) . Soon after that, a scrupulous analysis by the FDA and an independent group at Columbia University ultimately resulted in an FDA-mandated black box warning highlighting the potential for suicidal ideation in youths treated with SSRIs. The warning was later extended to all antidepressants
(5) . Since the black box warning was issued, the use of SSRIs in children has decreased
(6) and concern about treatment-emergent suicidal ideation has extended to adults.
The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial, the largest prospective treatment trial for major depressive disorder, provides a unique opportunity to ascertain treatment-emergent suicidal ideation prospectively in a large cohort of patients treated with the SSRI citalopram and to test whether specific genetic markers can identify patients who have an increased risk of developing this uncommon adverse event. Here we report an initial screen with 768 markers in 68 candidate genes. The results suggest that genetic markers may be able to identify some people at increased risk of treatment-emergent suicidal ideation. If replicated, these findings could have implications for the clinical management of major depression with SSRIs.
Results
Descriptive Variables
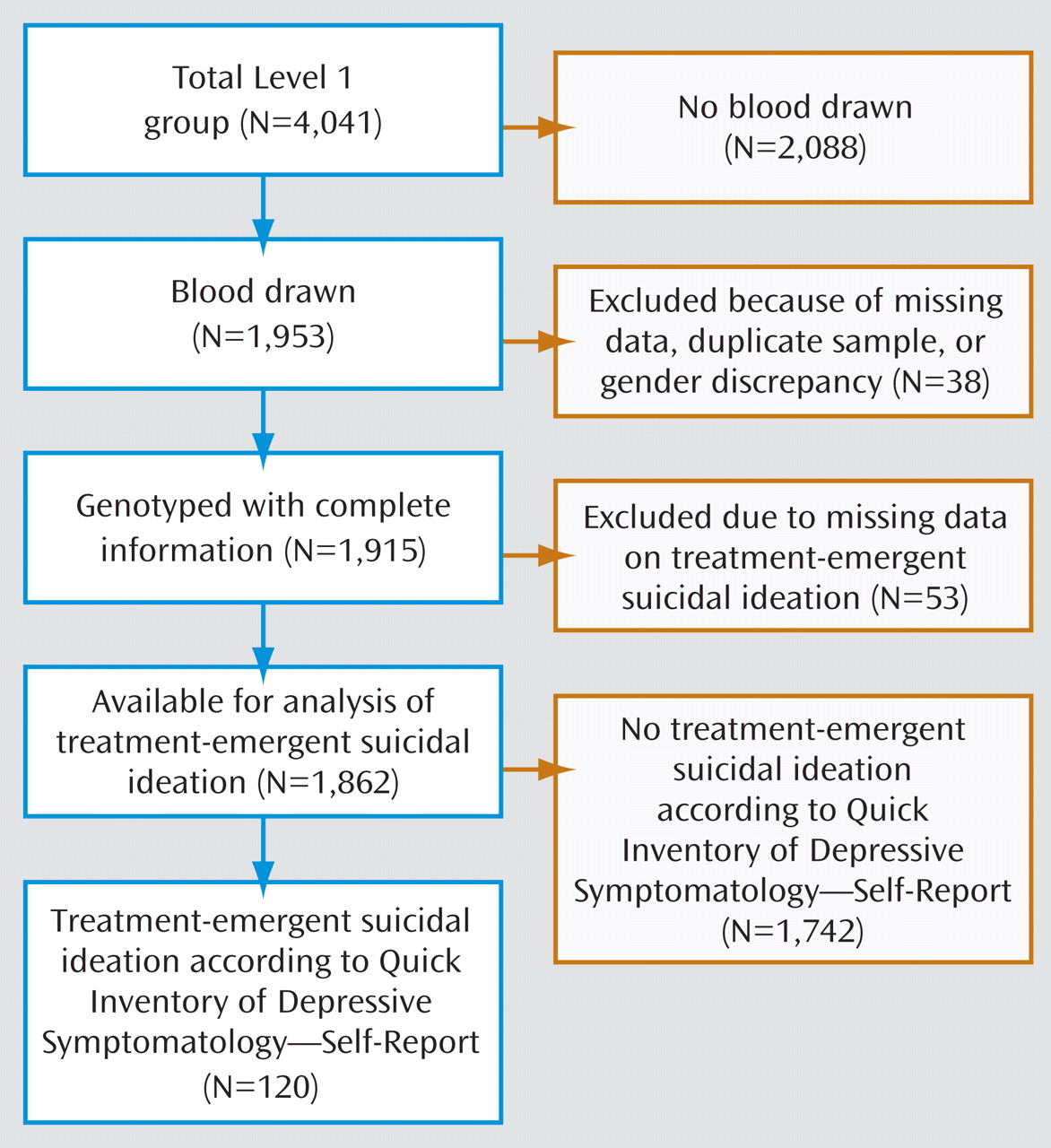
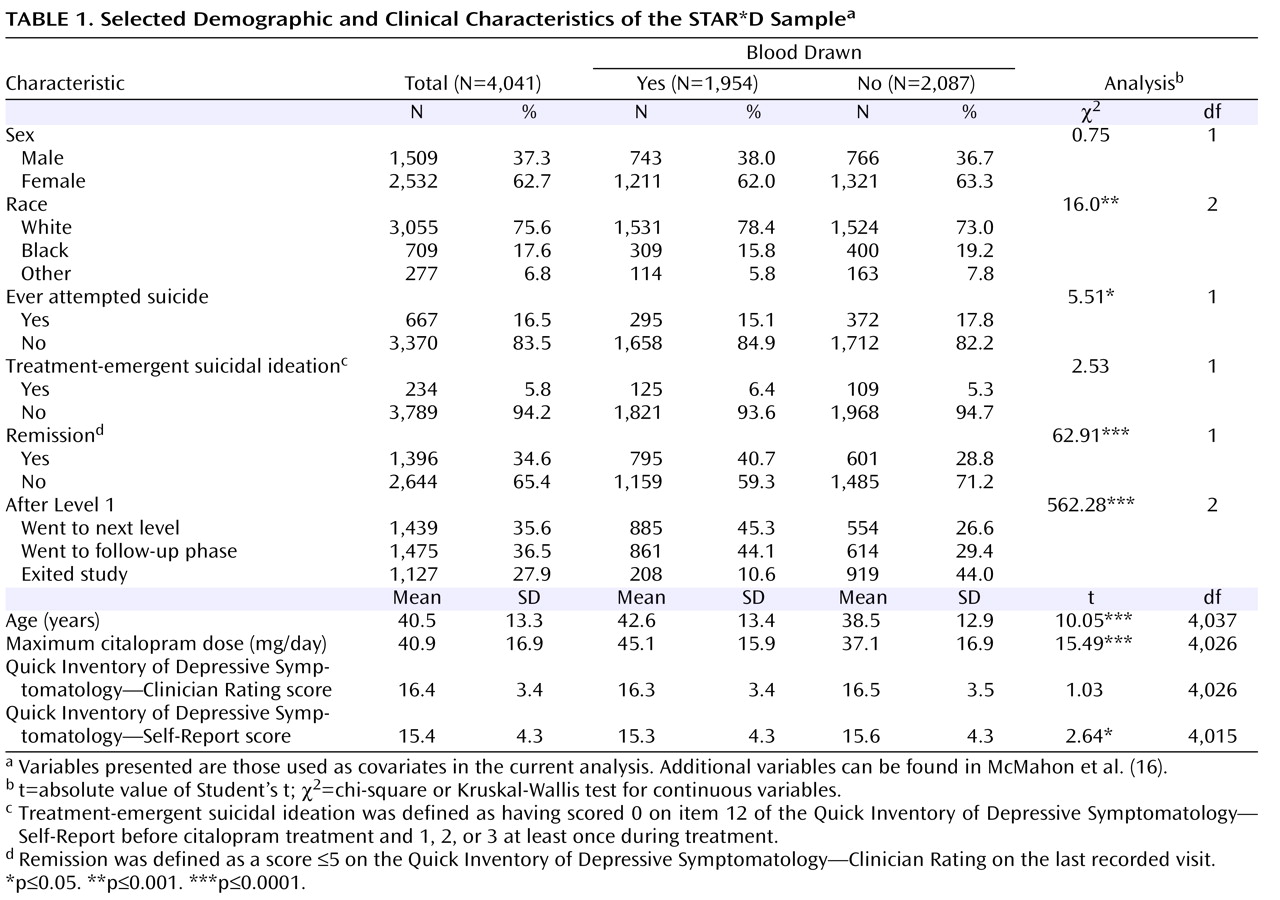
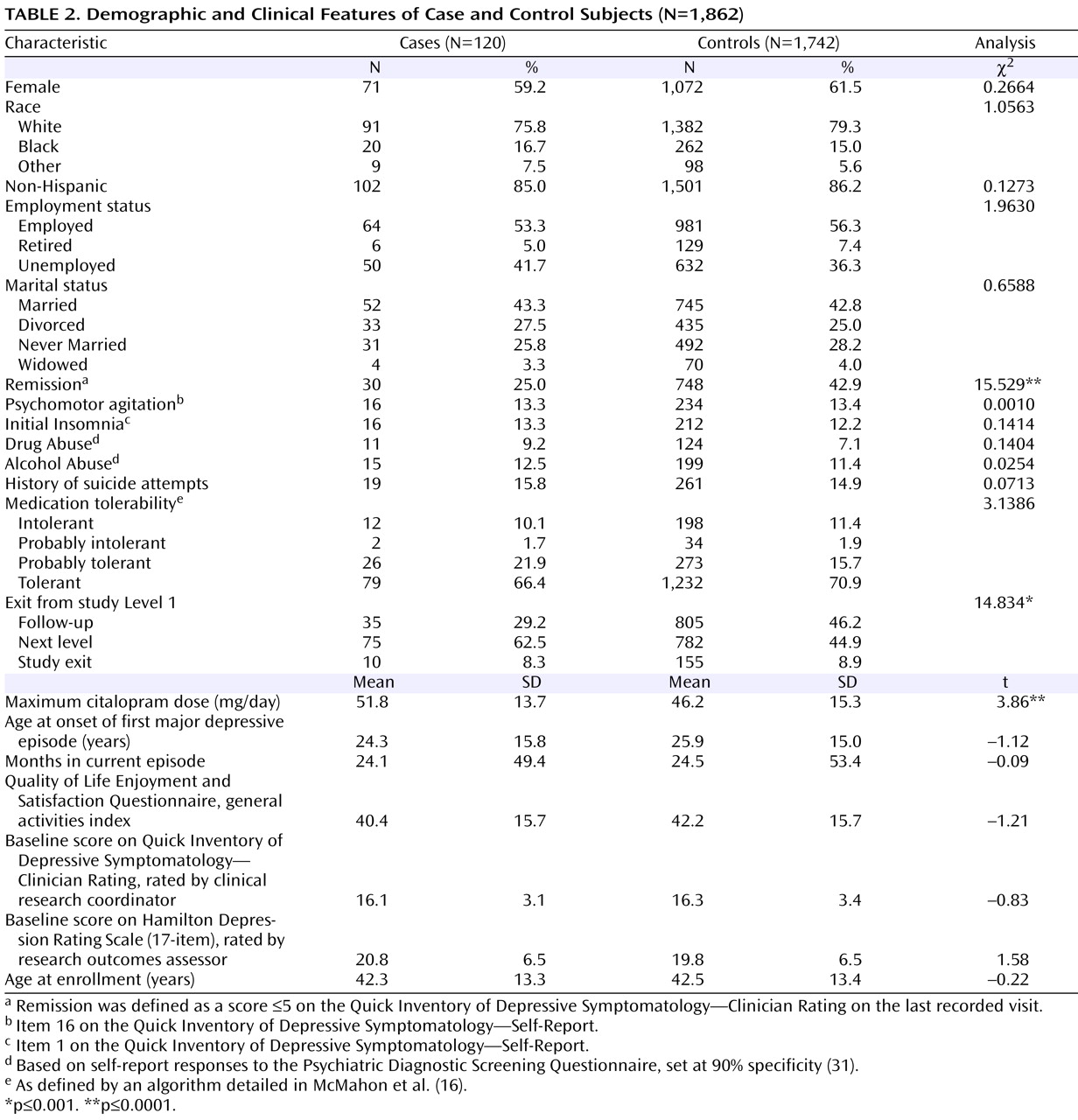
The demographic and clinical characteristics of the sample are presented in
Table 2 . There were no significant demographic differences between case and control subjects, and no differences in several clinical variables that are known predictors of suicide
(32) . Participants who developed suicidal ideation received a significantly higher maximum citalopram dose, were significantly less likely to go into remission (defined as a score £5 on the QIDS-C on the last recorded visit during citalopram treatment [Level 1]), and were more likely to move on to a secondary treatment phase (Level 2) after citalopram (15). Only 25% of those who had treatment-emergent suicidal ideation, compared with 42.9% of control subjects, achieved remission, although those with and without suicidal ideation did not differ significantly in initial symptom scores (
Table 1 ). Consistent with this poorer outcome, 62.5% of those with suicidal ideation went on to Level 2 to receive a change in treatment, compared with 44.9% of control subjects. There was no difference in the rates of exit from citalopram treatment.
Consistent with previous reports
(8), treatment-emergent suicidal ideation developed relatively early in treatment: 21% of those who developed suicidal ideation did so by visit 2 (median=14 days after starting treatment), 69% by visit 3 (median=21 days), and 92% by visit 5 (median=28 days). By the end of Level 1, 48% of those who developed suicidal ideation had returned to their baseline score of 0, while 37% persisted, and 15% had a fluctuating course. None of the participants with treatment-emergent suicidal ideation are known to have attempted suicide.
Association Analysis
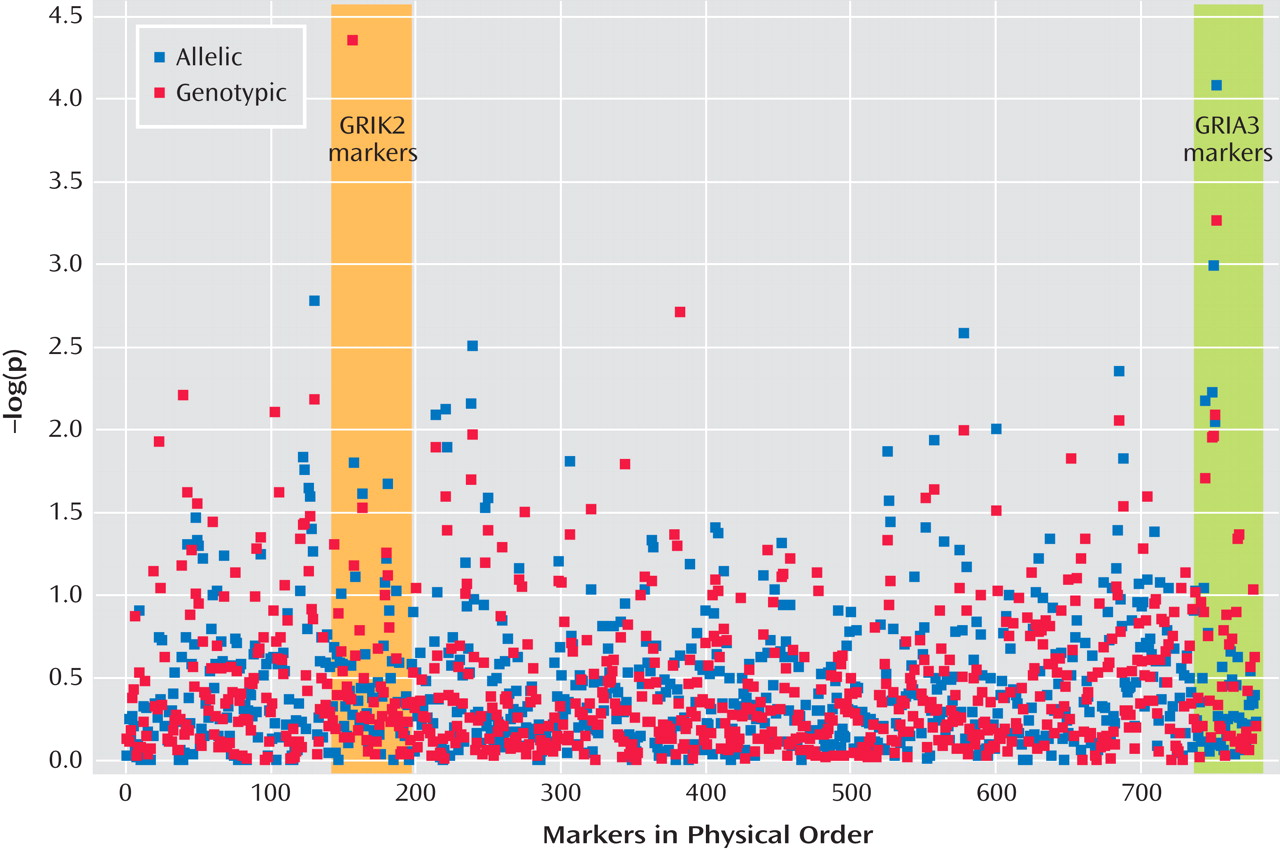
Figure 2 presents results of the allelic and genotypic association tests. Two markers produced significant evidence of association at the experimentwise p<0.05 level. Both markers were in Hardy-Weinberg equilibrium in this sample.
A marker in the first intron of GRIK2 on chromosome 6 (marker rs2518224), which encodes the kainate-sensitive ionotropic glutamate receptor GluR6, was associated with treatment-emergent suicidal ideation in the genotypic test (CC genotype, nominal p=2.43×10 –5, odds ratio=8.23; permutation p<0.003). This marker was not significantly associated in the allelic test.
A marker in the third intron of GRIA3 on chromosome X (marker rs4825476), which encodes the a-amino-5-hydroxy-3-methyl-4-isoxazole propionic acid-sensitive ionotropic glutamate receptor AMPA3, was associated with treatment-emergent suicidal ideation in the allelic test (G allele, nominal p=7.84×10 –5, odds ratio=1.94; permutation p<0.01).
Regression Analysis
Having established experimentwise significant associations between two markers and treatment-emergent suicidal ideation in this sample, we next investigated the impact of nongenetic variables on the observed genetic association. By means of stepwise logistic regression, we tested the impact of race and the three clinical variables that had shown significant differences in those with and without treatment-emergent suicidal ideation in our initial analysis (see
Table 2 ).
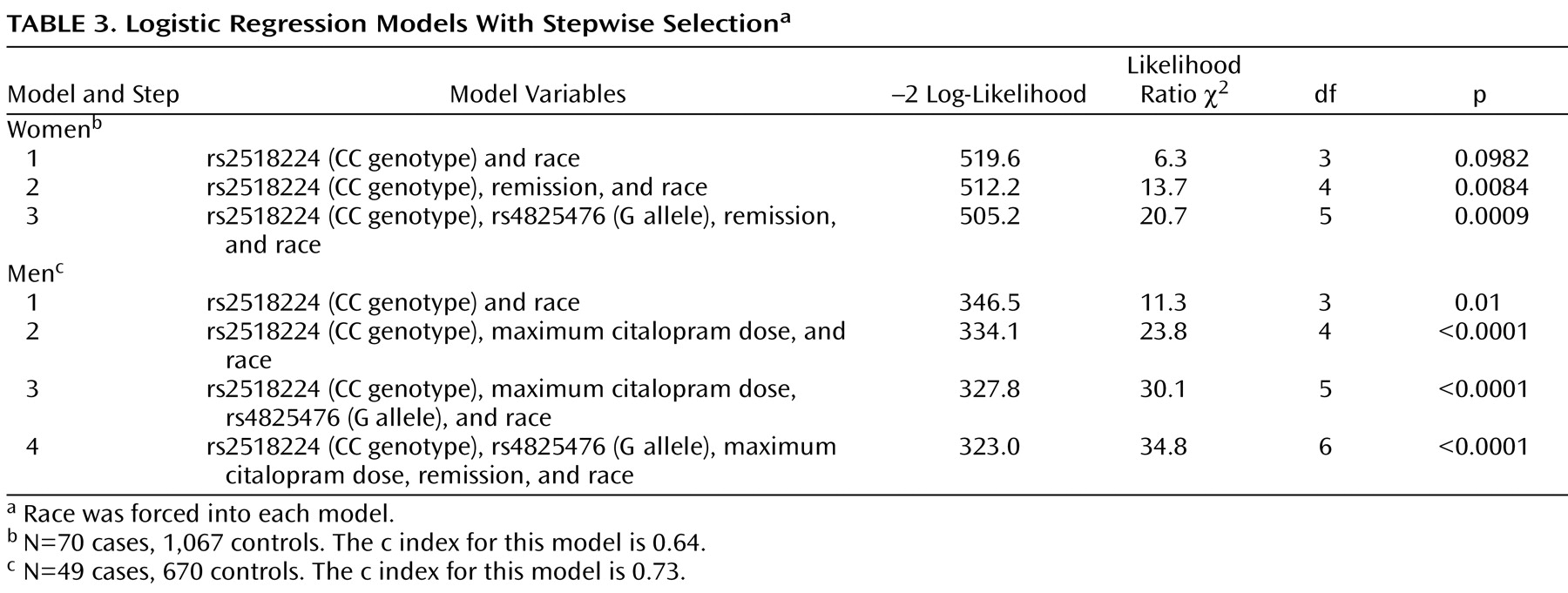
The best-fitting model was achieved with a combination of both markers, maximum citalopram dose, and remission by QIDS-C (
Table 3 ). Race was not a significant covariate in this model. Adjusted odds ratios were close to those in the unadjusted models. The Hosmer-Lemeshow test was nonsignificant for both genders, indicating a good model fit.
Population Structure
The association p values were uniformly distributed, with no excess of small values (Kolmogorov-Smirnov D=0.032, p=0.11). In the white subset, STRAT detected no evidence of mismatch between cases and controls (χ 2 =303.36, df=310, p=0.6).
Robustness to Varying Case Definition
The optimal case definition for treatment-emergent suicidal ideation is unknown. As a secondary analysis to assess the impact of varying case definition on the observed association findings, we examined treatment-emergent suicidal ideation as defined by the clinician-rated QIDS-C. The overall Pearson correlation of the QIDS-SR and QIDS-C scores on item 12 was low but highly significant (r=0.37, p=0.0001). The 144 cases of suicidal ideation defined by the QIDS-C alone showed association with markers in both GRIK2 and GRIA3 at the nominal p<0.01 level, although different single-nucleotide polymorphisms were involved. Participants who met the case definition for suicidal ideation on both the QIDS-SR and the QIDS-C (N=55) were significantly more likely to carry exactly the same marker alleles identified in our primary analysis than those who denied all suicidal thoughts on both instruments (χ 2 =15.42, df=2, p=0.0004).
Since our primary case definition included individuals who scored only 1 (“I feel that life is empty or wonder if it is worth living”) on the “thoughts of death or suicide” item, we compared allelic and genotypic frequencies of the GRIK2 and GRIA3 markers in those with treatment-emergent suicidal ideation who scored 1 and those who scored >1. There were no significant differences, which suggests that all patients who scored over 0 are similar with respect to allele frequencies at the GRIK2 and GRIA3 markers.
Combined Effect of Risk Alleles and Genotypes
Of the six combinations of high-risk alleles and genotypes tested, the highest odds ratio was observed in patients carrying both the high-risk allele of marker rs4825476 and the high-risk genotype of marker rs2518224 (odds ratio=14.98, 95% CI=3.7–60.674). Consistent with this, there was significant evidence of interaction between the two markers by the likelihood ratio test (χ 2 =12.3, df=1, p=0.0004). The combined impact of both markers on the risk of treatment-emergent suicidal ideation appears to be at least additive, but sample size limitations preclude any precise estimates of the mode of interaction.
Discussion
To our knowledge, this is the first study to detect a significant overall association between a genetic marker and treatment-emergent suicidal ideation. These data suggest that this uncommon but potentially dangerous adverse event may have a genetic component. Until functional alleles are demonstrated or replication is shown in an independent sample, these findings should be viewed as preliminary. However, they may shed light on the biological basis of suicidal ideation that emerges during antidepressant treatment and provide an initial step toward developing markers with clinically meaningful predictive value.
The validation of these findings through replication will be difficult, since treatment-emergent suicidal ideation is so uncommon
(9) . Several thousand patients may need to be studied before a sufficiently large replication sample can be obtained. To our knowledge, the STAR*D sample is the only such sample available at this time. However, the association findings we report here show several hallmarks of causal associations
(33) . The overall sample is large and is representative of major depression in the outpatient setting. The effect sizes are relatively large, and the statistical significance levels stand up to correction for the number of tests carried out. There is an apparent dose-response relationship between these markers and treatment-emergent suicidal ideation: more participants who carried both risk alleles reported suicidal ideation than those who carried only one allele. The implicated genes are biologically plausible candidates with closely related functions. Furthermore, association between these genes and treatment-emergent suicidal ideation persists across alternative case definitions.
This study has several limitations. Treatment-emergent suicidal ideation was defined on the basis of items in a depression rating scale, and neither the instrument nor the STAR*D study was designed to address this adverse event. Since there was no placebo group, we cannot determine what fraction of suicidal ideation in this study sample is directly attributable to antidepressant treatment. Moreover, the case definition we used is arbitrary. Given the lack of a widely accepted definition, we used one similar to that seen in the literature as well as that adopted by the FDA, which instituted the black box warning. Alternative case definitions are possible. We tested one alternative definition based on the QIDS-C, which yielded genetic association results similar to those we obtained with the definition based on QIDS-SR, and our results would remain significant at the p<0.05 level even if we doubled the permutation p values to account for a second possible phenotype definition. These limitations are characteristic of all existing large antidepressant treatment samples and highlight the need for large, placebo-controlled studies of treatment-emergent suicidal ideation in the future.
We observed that participants who developed suicidal ideation experienced a less favorable response to treatment overall, which could confound the interpretation of genetic association results. To address this limitation, we controlled for citalopram dose and remission rates in the secondary regression analysis. This analysis showed that the genetic markers we report were significant independent predictors of treatment-emergent suicidal ideation in this sample.
Another limitation of this particular study is that all participants were treated only with citalopram, so these findings may not be generalizable to other antidepressant medications. Citalopram is similar to other SSRIs and is one of the most widely prescribed antidepressants, so these findings have clinical relevance even if confined to citalopram and related compounds.
This study interrogated 68 genes. Although these genes were considered likely candidates for outcomes related to antidepressant treatment, there may be additional genes related to treatment-emergent suicidal ideation that were not studied here. For example, one previous study (34) found that markers in CREB1 were associated with treatment-emergent suicidal ideation in men. Moreover, the markers used did not cover the selected genes completely. Additional markers could detect additional association signals. Since markers were selected on the basis of low intermarker LD, haplotype tests would be expected to have poor power and thus were not performed. Still, this is the most comprehensive genetic study of treatment-emergent suicidal ideation conducted to date. More comprehensive studies will not likely emerge until after genome-wide association studies have been performed with this sample. As is true for most large studies, additional phenotypes have been and will continue to be tested over time in this sample. Our experiment-wide p value corrections, which are based on the hypotheses tested in this study, should be considered in this light.
Cryptic population stratification is always a risk in case-control genetic association studies. To address this, we showed that 1) there is no relationship between race and treatment-emergent suicidal ideation in this sample; 2) association p values were uniformly distributed within the largest (white) subset under the assumption of one population; and 3) there is no evidence of mismatch between participants with and without suicidal ideation within the white subset. Thus, these association results are not likely to be the result of population stratification.
While treatment-emergent suicidal ideation is an understandable cause for concern and has fueled widespread reassessment of antidepressant prescribing practices, it is not clear how it is related to suicidal behavior
(9) . Evidence suggests that the risk of suicide attempt is higher in depressed persons before the prescription of antidepressants than afterward
(35) . Only two participants attempted suicide while undergoing treatment in the STAR*D study, and one of them participated in DNA collection. Although this patient consistently denied suicidal ideation, he did carry the high-risk alleles for both GRIK2 and GRIA3.
The markers we have identified do not appear to be related to a general tendency toward suicide but rather to suicidal thoughts specifically emerging during antidepressant treatment. To our knowledge, neither of these genes has been previously associated with suicidal ideation. There was no difference in allele or genotype frequencies at either marker rs2518224 or marker rs4825476 in participants who had a history of suicide attempts or reported suicidal ideation at their initial visit before the start of treatment (data not shown).
Both markers implicated in this study lie within genes that encode ionotropic glutamate receptors. This is consistent with prior evidence that antidepressants affect glutamate signaling. Agonists to both ionotropic and metabotropic glutamate receptors may have an antidepressant-like effect
(36) . Chronic antidepressant treatment increases membrane expression of AMPA receptors in rat hippocampus
(37) in a time frame consistent with therapeutic effects. In addition, chronic treatment with SSRIs increases phosphorylation of the active sites of AMPA receptors in extracts of cortex, hippocampus, and striatum
(38) . A recent study demonstrated specific and regionally discrete changes in the expression and editing of AMPA and kainate glutamate receptors, along with selective reduction of conductance for GluR3-containing receptors, after treatment with antidepressants
(39) . That study also showed that prolonged exposure to antidepressants produced site-selective and area-specific effects on this particular posttranscriptional regulation.
In summary, we identified markers in two genes within the glutamate signaling pathway that may shed light on the biological basis of treatment-emergent suicidal ideation and have the potential to help identify patients at high risk of having suicidal ideation emerge during citalopram treatment. Such patients may benefit from closer monitoring, alternative treatments, or specialty care. Further work is needed to replicate these findings and uncover the functional variation that underlies the association signals we observed.