“J” was adopted at age 2 from a Romanian orphanage with no information regarding her family history or prenatal or perinatal course. She lacked functional language skills at the time of adoption and could neither crawl nor walk. Upon arrival in the U.S., she was found to be physically healthy, with a normal 46 XX karyotype. She participated in intensive developmental services with normalization of motor skills yet was diagnosed as autistic at age 3 owing to ongoing language, communication, and interpersonal impairments. Mental retardation was diagnosed at age 5.
As early as age 5, J showed sporadic “freezing” (her parents’ words) episodes in which her arm would suddenly shoot out and remain immobile for several seconds. She also developed both vocal and motor tics and was additionally diagnosed with Tourette’s syndrome. Clonidine and trials of antipsychotic drugs did not alleviate the tics, and they led to further behavioral deterioration.
J further developed multiple challenging behaviors, including yelling, screaming, and biting her lips and gums. These problem behaviors progressed to include hand-to-head, knee-to-head, and hand-to-body self-injury as well. By age 11, she had developed bilateral traumatic cataracts necessitating surgery and the introduction of protective equipment. She subsequently underwent years of unsuccessful psychotropic drug trials and intensive behavioral analysis.
At age 16, J acutely developed spastic movements that progressed to rigidity, staring, and lack of responsiveness to voice or touch. All investigations were normal in the emergency department, and the symptoms resolved completely with lorazepam. She was discharged with a diagnosis of acute dystonic reaction, although she had not been taking any medication, psychotropic or otherwise.
Five months later, J required surgery due to traumatic heterotopic ossifications in her thigh from chronic self-injury. While eating a hamburger postoperatively, she became unresponsive, stopped eating, held her mouth agape and stared for several minutes. This presentation abated over the course of an hour and was attributed to an abnormal reaction to pain medication. J did well for the next few months, with the exception of the staring and freezing episodes. These could happen during any activity; her mother remembers asking J to throw out the trash, then seeing J suddenly stop over the garbage can with a frozen expression and her hand suspended in the air, clutching the trash for several minutes before releasing it.
Over the next months, J’s self-injury increased to hundreds of times hourly, and she developed choreiform movements of her hips and shoulders. J completely stopped eating and showed alternating immobility and frenzied physical agitation. She was hospitalized and diagnosed with catatonia. A trial of lorazepam, 2 mg intravenously every 2 hours, afforded some initial symptom resolution. Neither ongoing lorazepam treatment nor ECT was pursued, reportedly due to her developmental disability. J underwent gastric tube placement and was sent home with a helmet and in four-point restraints.
J’s catatonia and self-injury subsequently worsened before admission to our neurobehavioral unit. J relied entirely on her gastric tube for nutrition and spent the majority of the day immobile, posturing, staring, and mute. Of interest, during the 11-hour ride to the facility, J received 2 mg of clonazepam in four evenly divided doses. Her parents reported a lack of self-injury and posturing during the trip, as well as a sudden clear request to stop at a fast food restaurant and purchase a roast beef sandwich, which J ate in its entirety.
On admission, J was an emaciated, disheveled young woman wearing a helmet and bilateral arm restraints and staring at the floor. She did not respond to greetings nor did she look at the new surroundings or staff. She required physical guidance to walk onto the unit and demonstrated a wide-based, shuffling gait. She then sat immobile with her arms outstretched and continued staring at the floor. She did not interact with her parents or staff and uttered only an occasional grunt. From the first days of admission, she was noted to engage in various postures involving her arms, fingers, torso, and legs sustained for several minutes and resisting repositioning ( Figure 1 ).
Figure 1. J Posturing in a Jackknife Position at Age 18
Method
Our working diagnosis was catatonia since J met all DSM-IV-TR criteria with the exception of echolalia or echopraxia. Medical and neurological causes of the catatonia were eliminated with comprehensive laboratory testing, brain magnetic resonance imaging (MRI), and EEG. We considered a comorbid mood or schizophrenic illness; however, the severity of her presentation made it difficult for us to be conclusive about such. Although her presentation could have been consistent with depression, she had previously experienced complete remission of all symptoms with administration of lorazepam. Moreover, there was no history of depressive symptoms when catatonia was absent. At admission, we also administered a lorazepam challenge, with dosages up to 8 mg/day, resulting in increasing alertness, interactions, toy play, verbalizations, and appetite with decreased posturing. In fact, after her first lorazepam dose, she suddenly asked to eat chicken.
Nevertheless, to cover a possible underlying depressive illness, we started a regimen of citalopram ultimately titrated to 35 mg/day. Neuroleptics were avoided owing to the risk of worsening the catatonia (1) . Lorazepam was maintained during the 3 weeks required to gain legal authorization for ECT, yet benefit rapidly waned, and ECT was pursued as a definitive treatment.
Modified ECT was supervised by I.M.R. and administered with a MECTA Spectrum 5000Q (Mecta Corporation, Tualatin, Ore.) unit. For each treatment, anesthesia was induced with methohexital, 70 mg intravenously, and succinylcholine, 60 mg intravenously, was administered for muscle relaxation. Seizure activity was monitored clinically and by EEG with bifrontal electrode placement. Recorded seizure length was ascertained by EEG. For the first round of ECT treatment, before the partial retinal detachment, electrode placement was right unilateral. There were 12 treatments administered three times per week with a starting dose of 128 millicoloumbs (mC) and an average dose of 305 mC. The average seizure length was 46 seconds.
Results
After her first three ECT treatments (ECT 1), J made eye contact upon greeting and responded with “Hi.” She also began assessing her surroundings by looking at peers and staff rather than staring. After four ECT treatments, J spontaneously asked staff, “How are you?” and made other brief comments. She also gave her name in group activities. After ECT number 7, posturing was significantly reduced, J used full sentences to make requests, and she showed an ongoing reduction in negativism. By ECT number 9, posturing was absent, oral intake was greater than 90% of the meals presented, sleep was stabilized at 9 hours nightly, and J was freely verbalizing, interacting, and playing with staff. Self-injury also showed marked reduction.
Unfortunately, after 12 ECT treatments, J was found to have a detached left retina from old self-injury before ECT. She underwent laser retinal reattachment with oil bubble and scleral buckle procedures, and ECT was postponed for 6 weeks due to potential increased intraocular pressure during ECT and the risk for oil and suture disruption. Exactly 10 days after her last ECT treatment, posturing resumed in J’s upper extremities. Full catatonic deterioration ensued rapidly.
ECT (ECT 2) recommenced 6 weeks postoperatively, and a positive effect was again seen after the first four treatments. During the second course, J received 13 right unilateral treatments, three times per week, before tapering to maintenance. For the second course, the initial dose was 160 mC, the average dose was 374 mC, and the average seizure length was 49 seconds. The treatment taper began with right unilateral administration two times per week at maximal charge, i.e., 576 mC. However, because J showed breakthrough signs of catatonia on the 5th day after a right unilateral treatment, she was switched to bilateral lead placement, having received a total of five right unilateral treatments during the first part of the taper. Bilateral treatment dose was initially 432 mC, and treatment frequency was increased to three times per week until all symptoms had resolved. At the time of the writing of this article, treatment was again twice weekly, with further tapering to be continued with J as an outpatient.
With ECT, J’s catatonic stupor resolved, and she returned to her baseline 3- to 4-year-old functioning level. She spoke for interaction as well as to communicate her needs, was able to toilet and feed herself, laughed, smiled, danced, played, sang, and once again safely enjoyed activities in the community, all accompanied by the near eradication of self-injury.
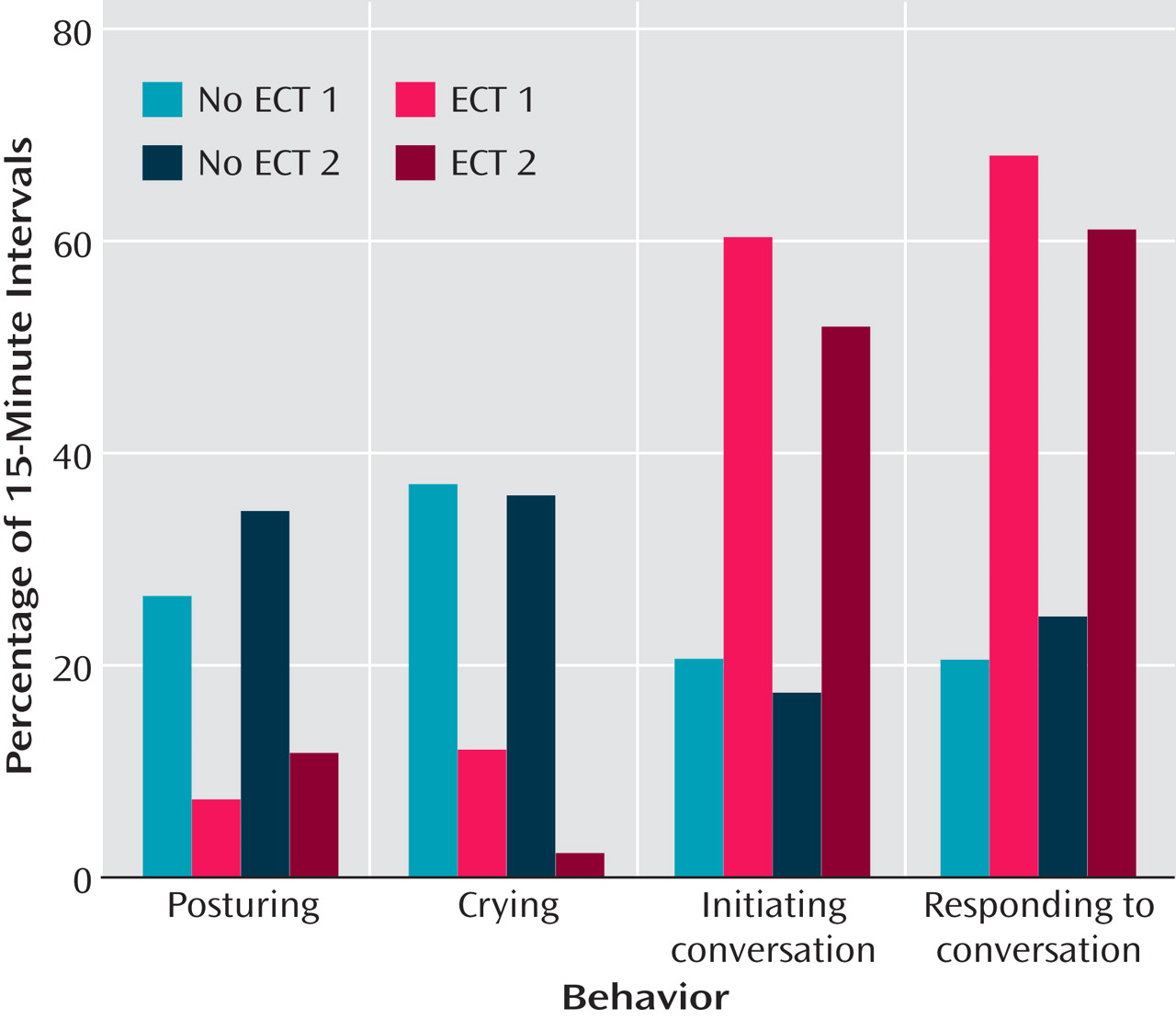
Figure 2 shows average percentage of 15-minute intervals of her posturing, crying, initiating conversation, and responding to conversation with and without ECT (the last 14 days of each condition). A partial interval data collection procedure was used. Therefore, data were recorded regarding whether or not J engaged in that behavior during the 15-minute observation interval. This was done during all waking hours. The first ECT phase (ECT 1) resulted in marked improvements in all behaviors. Posturing and crying decreased by 73.5% and 67.9% (compared to no ECT 1), respectively. Initiating and responding to conversations increased by 296.6% and 334.5%, respectively. Similar improvements in these behaviors were observed after the second ECT phase (ECT 2). Posturing and crying decreased by 66.5% and 94.7% (compared to no ECT 2), respectively. Initiating and responding to conversations increased by 297.1% and 247.0%, respectively.
Figure 2. J’s Data for Posturing and Crying as Well as Initiating and Responding to Conversation From the Last 14 Days of Each Phase (no ECT versus ECT)
Discussion
Catatonia in Autism Spectrum Disorders
The concomitance of catatonia and autism spectrum disorders has received increased interest and attention in recent years. Although catatonia has historically been associated with schizophrenia, research in psychiatric inpatients has found catatonia most frequently associated with mood disorders, particularly bipolar disorder (1) . Investigations into patients with autism spectrum disorders have demonstrated a high rate of catatonic features, with frank diagnosable catatonia in 11%–17% of cases (2 – 4) . The relationship between autism and catatonia has been further explored in case studies and reports (5 – 8), and it is currently postulated that both processes may share a common neuronal substrate as well as a shared genetic susceptibility region on chromosome 15q (9, 10) .
This diagnostic concomitance is of both historical and current import. Catatonia was originally described by Karl Kahlbaum in 1874 as a constellation of motor, affective, and vocal symptoms occurring in all age groups, including children (11) . Presently, catatonia is only characterized by DSM-IV-TR as a specifier for schizophrenia, primary mood disorders, and mental disorders due to a general medical condition. These criteria require at least two of five symptoms, including 1) motoric immobility, 2) excessive motor activity, 3) extreme negativism, 4) peculiarities of voluntary movement, and 5) echolalia or echopraxia (12) . Many modern researchers believe catatonia may actually represent a separate neurobiological syndrome (1, 13) .
The term “autism” was first introduced by Bleuler in 1910 in reference to schizophrenic negativism and withdrawal yet was established as a separate psychiatric syndrome in 1943 by Kanner (14) . The DSM-IV-TR criteria for autism demand core dysfunction in 1) social interaction and 2) communication as well as 3) restricted or stereotyped patterns of behavior, interest, and activity.
Like catatonia, specific DSM symptoms for autism may include unusual mannerisms, gestures, and postures; mutism; echolalia or stereotypic speech; and stereotypic or repetitive motor behaviors. Wing and Shah (3, 4) suggested expanded criteria for the diagnosis of catatonia in autistic individuals, including eight symptom clusters: 1) slowed movements and verbalizations, 2) slowed task initiation and completion, 3) reliance on prompting, 4) passivity/amotivation, 5) parkinsonian features, 6) day-night reversal, 7) repetitive/ritualistic behavior, and 8) agitation/excitement. There is also suggestion that catatonia found in autism may represent a separate clinical phenomenon (15) .
The clinical overlap between the two syndromes is evident. In three separate studies of children and adults with autism (3, 4), Wing and Shah found catatonic deterioration in 17%. They asserted that catatonic stupor interfering with activities of daily life was indeed rare, with insidious onset and rarely appearing before adolescence or early adulthood. Like J, many of these patients came to clinical attention because of challenging behaviors. Wing and Shah further speculated that baseline catatonia-like features in autistic individuals render them more vulnerable to later catatonic deterioration.
In a population-based study of patients with autism diagnosed in childhood (2), 13 (12%) autistic individuals between ages 17 and 40 years had clinically diagnosable catatonia with severe motor initiation problems. An additional four individuals had several catatonic symptoms but not the full syndrome.
In another report on 11 autistic and catatonic adult males (16), Ohta et al. found the onset of catatonia at an average age of 19 years, with a history of slowed movements before catatonic deterioration in eight of 11—one patient demonstrating motor immobility as early as 15 months—the presence of Tourette’s syndrome in three of 11 subjects, and increased self-injury in two of 11. Fink et al. (17) similarly describe four 17-year-old boys with autism spectrum disorder and catatonic deterioration, all of whom showed insidious onset of symptoms with motor changes months to years before frank catatonia, as well as frequent concomitance of new-onset tics. These findings all resonate with J’s history, in which motor episodes and tics were present from early childhood and self-injury was lifelong yet intensified during catatonic deterioration. The question is also raised as to the potential links between autism, catatonia, self-injury, and tic disorders in that these may all share a common substrate.
It has indeed been proposed that catatonia and autism share common neuronal dysfunction with differences in age of onset accounting for the incomplete symptom overlap (8, 9) . Genetic linkage studies have found a shared suspectibility region, 15q15-q21, for both autism and catatonia (10) . Genes in this region are believed to encode for g-aminobutyric acid (GABA) receptors B3, A5, and G3, which is of further interest because of the use of benzodiazepines in catatonia as well as the role of GABA in ECT (18) .
ECT Responsiveness
Regarding treatment, J showed a resounding response to ECT. ECT has been successfully used to treat catatonia since the 1930s (1, 19) and is indicated for patients who do not respond to a trial of benzodiazepines. The mechanism by which ECT is effective for catatonia is not understood, although several theories have been proposed (18, 20, 21) . One theory argues that reversal of neuroendocrine dysregulation through hormonal discharge from the hypothalamus following induced seizures is critical for the effectiveness of ECT in catatonia and other severe psychiatric illnesses (21) . Other theories concern the therapeutic effects of ECT on various putative abnormalities in catatonia, such as GABA dysfunction, disturbed neurogenesis in critical brain areas, or excess frontal lobe activity. A recent review (21) emphasized that “a sustained search for why induced seizures relieve mood disorders is overdue.” Similar studies are warranted for catatonia that is shown in this case report to occur in people with autism and to respond to ECT.
To treat J, we started with right unilateral placement to minimize cognitive side effects as is usually the practice at Johns Hopkins University Hospital. However, a switch to bilateral placement was necessitated by breakthrough catatonic symptoms during treatment taper. This lends support to the proposal that catatonia should be treated from the outset with bilateral ECT (1) . Although ECT is clearly efficacious as maintenance treatment for patients with depressive disorders who respond acutely and have failed to respond to pharmacotherapy, no data exist on the use of maintenance ECT for catatonia. Nevertheless, because reports of harm related to ECT in children and adolescents are exceedingly rare (22), and there are no reports of harm associated with maintenance ECT treatment, we are recommending gradual tapering to maintenance treatment as was done for this patient. Dhossche et al. (23) recommended gradual tapering of maintenance treatment over at least 3 to 6 months in autistic patients recovering from catatonia. Since J responded quickly to ECT during both acute courses, it is likely she would respond quickly if symptoms reemerge during tapering and maintenance.
Finally, the treatment modality of ECT itself deserves mention. The use of ECT is fraught with controversy in the United States and abroad, with public interest groups comparing ECT providers to sadists and often affecting state laws regarding ECT. As more children are diagnosed with autism, it is likely that more such youngsters will be later diagnosed with catatonia. These patients may have significant difficulty receiving ECT in the states with severe restrictions on pediatric ECT. Although the legal regulations for J’s treatment in the state of Maryland required only that a guardian be court appointed, significant legal and administrative barriers to ECT exist in many states, often precluding or severely restricting prompt access to desperately needed treatment. These barriers are frequently amplified in the case of juveniles or patients with cognitive impairment, particularly related to concerns of informed consent. This situation persists (24, 25) despite clear indications for ECT issued by APA in a variety of psychiatric conditions, including catatonia. In mentally retarded patients with concomitant psychiatric illness, ECT has historically been rarely used, yet several reports support the safe and efficacious use of ECT in this population (26 – 29) . Such most certainly was the case for J.
Conclusions
Recent reports indicate that individuals with autism spectrum disorders have an increased incidence of catatonic symptoms, as well as frank catatonic deterioration. This case report adds support to the limited literature that ECT is a highly effective and potentially life-saving treatment for catatonia in the general population and should figure prominently in the treatment algorithm for catatonic autistic patients as well. A further clinical implication is that autism should be considered in the differential diagnosis as a possible underlying condition in patients seen with catatonia.
Controlled studies in this area are needed. There are many diagnostic and clinical similarities between the two disorders, begging the question if there are common biological substrates. This issue also deserves more focused research.
Footnotes
Presented in part at the Distinguished Lecture Series, the Marcus Institute, Atlanta, July 11, 2007. Received Aug. 6, 2007; revision received Oct. 30, 2007; accepted Nov. 14, 2007 (doi: 10.1176/appi.ajp.2007.07081246). From the Departments of Psychiatry, Kennedy Krieger Institute, and Johns Hopkins University Hospital, Baltimore; and the University of Mississippi Medical Center, Jackson, Miss. Address correspondence and reprint requests to Dr. Wachtel, Kennedy Krieger Institute, 707 North Broadway St., Rm. 232, Baltimore, MD 21205; [email protected] (e-mail).
Dr. Cascella has been on the speakers bureaus of AstraZeneca, Eli Lilly, and Bristol-Myers Squibb and has received research grants from Astellas, Sanofi-Aventis, and Acadia. The remaining authors report no competing interests.
The authors thank J’s family for agreeing to share her case.
References
1.
Fink M, Taylor M: Catatonia: A Clinician’s Guide to Diagnosis and Treatment. Cambridge, UK, University Press, 2003
Billstedt E, Gillberg IC, Gillberg C: Autism after adolescence: population-based 13- to 22-year follow-up study of 120 individuals with autism diagnosed in childhood. J Autism Dev Disord 2005; 35:351–360; correction, 37:1822
Dhossche D, Wachtel LE: Catatonia in psychiatric illnesses, in The Medical Basis of Psychiatry, 3rd ed. Edited by Fatemi SH, Clayton P. Totowa, NJ, Humana Press (in press)
Ohta M, Kano Y, Nagai Y: Catatonia in individuals with autism spectrum disorders in adolescence and early adulthood: a long-term prospective study. Int Rev Neurobiol 2006; 72:41–54
Dhossche D, Rout U: Are autistic and catatonic regression related? a few working hypotheses involving GABA, Purkinje cell survival, neurogenesis, and ECT. Int Rev Neurobiol 2006; 72:55–79
Dhossche D, Shah A, Wing L: Blueprints for the assessment, treatment, and future study of catatonia in autism spectrum disorders. Int Rev Neurobiol 2006; 72:267–284
Winslade WJ, Liston EH, Ross JW, Weber KD: Medical, judicial, and statutory regulation of ECT in the United States. Am J Psychiatry 1984; 141:1349–1355
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).
If the address matches an existing account you will receive an email with instructions to retrieve your username
Create a new account
Change Password
Password Changed Successfully
Your password has been changed
Login
Reset password
Can't sign in? Forgot your password?
Enter your email address below and we will send you the reset instructions
If the address matches an existing account you will receive an email with instructions to reset your password.
Change Password
Congrats!
Your Phone has been verified
×
As described within the American Psychiatric Association (APA)'s Privacy Policy and Terms of Use, this website utilizes cookies, including for the purpose of offering an optimal online experience and services tailored to your preferences. Please read the entire Privacy Policy and Terms of Use. By closing this message, browsing this website, continuing the navigation, or otherwise continuing to use the APA's websites, you confirm that you understand and accept the terms of the Privacy Policy and Terms of Use, including the utilization of cookies.