The pathophysiology of delirium is associated with a wide variety of etiologies. Determining the etiology, if possible, is always the first step in managing the condition, and treating the underlying cause may be curative. The postoperative state may itself lead to delirium as the ebb phase of anesthetic-induced vasodilation precipitates a hypermetabolic inflammatory state that is proportional to the degree of surgical injury. The timeline of this phasic response is relatively immutable and coincides with the clinical presentation and course of postoperative delirium. The treatment of choice for the agitated behavior of postoperative delirium is neuroleptic medication, in most cases intravenous haloperidol, especially when agitation is severe. In this article, we offer a review of the diagnosis, risk factors, etiology, pathophysiology, management, and treatment of delirium.
Differential Diagnosis
Delirium is a state of acute brain failure with disturbance of consciousness accompanied by cognitive deficits that cannot be accounted for by past or evolving dementia and is associated with evidence of physiological disturbance owing to a medical condition
(9) . The DSM-IV-TR diagnostic criteria include:
A. Disturbance of consciousness (i.e., reduced clarity of awareness of the environment) with reduced ability to focus, sustain, or shift attention.
B. A change in cognition (such as memory deficit, disorientation, language disturbance) or the development of a perceptual disturbance that is not better accounted for by a preexisting, established, or evolving dementia.
C. The disturbance develops over a short period of time (usually hours to days) and tends to fluctuate during the course of the day.
Other common features of delirium include sleep disturbances (including reversal of the sleep-wake cycle), psychomotor activity changes, and neurobehavioral symptoms.
Hyperactive delirium is characterized by a state of hyperarousal, hypervigilance, and pronounced agitation. Patients with hypoactive delirium are hypoalert and lethargic; often their symptoms are not appreciated because of their nondisruptive nature. Occasionally, patients will have a delirium with features of both motoric types.
Labile or altered affective changes, such as anger and irritability as well as apathy and abulia, are frequently described. Patients may experience symptoms of psychosis, interweaving delusional and hallucinated events with real ones. Formal thought disorder may also be observed. When patients do not show autonomic derangements or perceptual changes, the moniker “acute confusional state” is sometimes preferred.
The differential diagnosis of delirium requires specific attention to symptom profile; temporal onset; physical, neurological, and mental status examinations; and results of radiological and laboratory testing
(10) . Asterixis and multifocal myoclonus are physical signs. In most non-alcohol-withdrawal deliria, EEG shows generalized slowing and increased amplitude
(11) . The presence of triphasic waves may be helpful in distinguishing certain specific metabolic deliria (e.g., hepatic encephalopathy). Abnormal clock drawing and dysgraphia are useful bedside findings.
Acute alcohol withdrawal is often seen in the inpatient setting, especially after surgery. Symptoms, generally due to adrenergic excess, include tremulousness, anxiety, diaphoresis, palpitations, and insomnia. When more pronounced, there may be hallucinosis, withdrawal seizures, and alcohol withdrawal syndrome or delirium tremens, which nowadays carries a mortality rate of 1%
(12) . Classically, alcohol withdrawal syndrome presents with fever, tachycardia, hypertension, tremor, insomnia, hallucinations (predominantly visual, but also auditory and tactile), agitation, anxiety, and seizures, along with reduced attention, disorientation, cognitive dysfunction, and language disturbances. Distinguishing alcohol withdrawal syndrome from postoperative delirium is a common challenge because of symptom overlap.
Dementia can be difficult to distinguish from delirium. Late-stage dementia can lead to confusion, agitation, and psychosis, and any type of cognitive impairment may predispose to delirium. Delirium superimposed on dementia, or “beclouded dementia,” has a prevalence ranging from 23% to 89% in elderly patients with dementia
(13) .
An accurate longitudinal history can help distinguish delirium from dementia. Abrupt onset of symptoms, with a waxing and waning course, is highly characteristic of delirium. Attention is significantly disturbed or fluctuates rapidly, whereas it remains relatively intact in uncomplicated dementia
(14) . Hallucinations, delusions, and agitation levels are often prominent in delirium but usually are absent in dementia until the late stages.
Hypoactive delirium is often misdiagnosed as depression, as some overlap of symptoms occurs. These include psychomotor slowing, cognitive changes, sleep disturbances, irritability, and in cases of depression with psychotic features, perceptual changes and delusions. Attention and orientation are usually normal in depressive disorders, however.
Agitation in hyperactive delirium is often related to fear either caused or accompanied by symptoms of psychosis. This is often misdiagnosed as anxiety disorder and mistreated with benzodiazepines. Perceptual disturbances, along with thought abnormalities, can occur in both delirium and primary psychotic disorders. However, symptoms in delirium are more fluctuant and fragmentary and rarely as fixed or complex. Hallucinations in delirium tend to be visual rather than auditory. In addition, consciousness, attention, and cognition are generally less impaired in primary psychoses.
While the clinical evaluation remains paramount—and often it is the only approach available when encountering serious agitation—the history and examination can occasionally be supplemented by use of formal instruments. The Confusion Rating Scale
(15), the Mini-Mental State Examination
(16), the clock drawing test
(17), and the Glasgow Coma Scale
(18) can be useful screens. The Delirium Symptom Interview, the Confusion Assessment Method
(19), and the Delirium Scale
(20) can all aid in formal diagnosis. Instruments used to rate delirium severity include the Delirium Rating Scale
(21) and the Memorial Delirium Assessment Scale
(22) .
Our patient exhibited classic symptoms and signs of a hyperactive postoperative delirium with clear onset on postoperative day 2. An alcohol withdrawal syndrome was initially suspected, but a blood alcohol level of 0, a negative CAGE screen, normal MCV, and normal levels of liver enzymes, bilirubin, and albumin argued against it. The patient’s non-small-cell carcinoma of the lung suggested the remote possibility of paraneoplastic limbic encephalitis, but his MRI was unremarkable and his postoperative delirium responded briskly to haloperidol.
Etiology and Pathophysiology
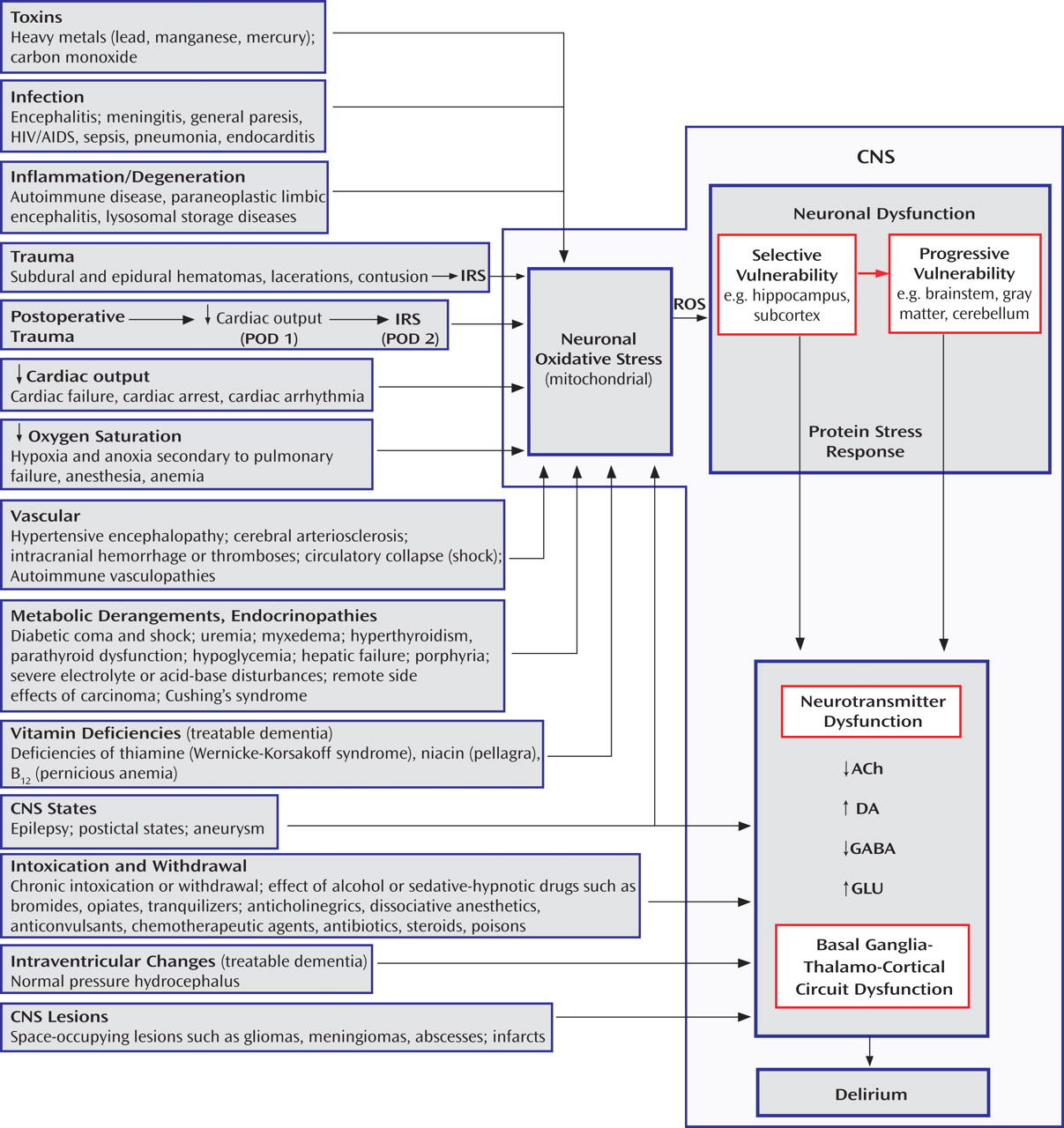
The pathophysiology of delirium is associated with a wide variety of etiologies (
Figure 1 ). Seven states (designated by the mnemonic WHHHIMP
[31] ) require prompt intervention to avoid death or permanent CNS damage: Wernicke’s encephalopathy, hypoxia, hypoglycemia, hypertensive encephalopathy, intracerebral hemorrhage, meningitis/encephalitis, and poisoning, whether exogenous or iatrogenic. Other conditions requiring prompt intervention include subdural hematoma, septicemia, subacute bacterial endocarditis, hepatic or renal failure, thyrotoxicosis/myxedema, alcohol withdrawal syndrome, anticholinergic psychosis, and complex partial status epilepticus. Postoperative delirium, which may be complicated by these conditions, bears a temporal and etiological relationship to surgery, usually developing between postoperative days 2 and 7.
Delirium is a potentially reversible dysregulation of neuronal functioning caused by oxidative stress initiated by a disturbance in brain structures most vulnerable to insult
(32) . This “selective vulnerability” results in dysfunction of specific neurotransmitter systems. If the insult persists, less vulnerable brain structures also become dysfunctional in a process called “progressive vulnerability.” Hippocampal neurons are affected early in delirium, followed by neurons of the subcortex, brainstem, gray matter, and cerebellum
(32) .
The inflammatory response syndrome resulting from anesthetized surgical or unanesthetized traumatic injury is a predictable phasic adaptation that optimizes the body’s healing capacity
(33) . The “ebb” phase is akin to shock, affecting cardiac output, heart rate, oxygen consumption, gluconeogenesis, lipolysis, and cytokine and catecholamine levels. This roughly 24-hour period represents an attempt to survive in the face of acute injury by decreasing energy requirements. This first phase occurs during emergencies of hemorrhage, sepsis, and dehydration, as well as after elective surgery, when common anesthetic agents profoundly reduce cardiac output, thus activating the conservatory mechanisms described above.
When surgical interventions expand circulatory volume, other survival signals are activated, initiating the second phase of “flow” or hypermetabolism. The degree of initial injury proportionately propels the hypermetabolic response, yet the timeline of the response is relatively immutable. On postoperative day 2, almost all of the inflammatory and catabolic mediators peak, and they return to baseline levels by days 6 to 7. This course is closely correlated with the clinical course of an uncomplicated postoperative delirium, which suggests that the inflammatory response syndrome may be pathophysiologically involved.
For example, the inflammatory response syndrome may then affect circumventricular fenestrated endothelial zones, creating areas of inflammation leading to limbic disturbance and symptoms of delirium
(34) . The transcription factor NF-kB is excited by the inflammatory response syndrome as well as by both oxidative and psychosocial stress, resulting in proinflammatory cytokine stimulation of delirium-potentiating neurotoxins
(35) . An inflammatory response syndrome-stimulated macrophage enzyme called indoleamine 2,3-dioxygenase favors kynurenine production
(36) . Excess kynurenine crossing the blood-brain barrier will potentiate a delirium through oxidative stress. In addition, the inflammatory response syndrome stimulates excessive cortisol release, which can exacerbate delirium.
Cholinergic, dopaminergic, histaminergic, noradrenergic, and serotonergic neurons are most susceptible to oxidative stress. Oxidative stress leads to neuronal calcium influx, mitochondrial strain, and a protein stress response. Enhanced release of some, but not all, neurotransmitters occurs, because while some enzymatic reactions require oxygen as a cofactor, others do not. For example, glutamate increases during hypoxia, which causes excitotoxicity, whereas oxidative stress decreases the synthesis of γ-aminobutyric acid (GABA), and even mild hypoxia interferes with a cholinergic cell’s ability to synthesize and release acetylcholine
(32,
37) .
In delirium, there is dysfunction of several brain nodal points, including subcortical structures such as the amygdala and hippocampus, the brainstem, the thalamus, the nondominant parietal lobe, and the prefrontal cortices, all connected by basal ganglia-thalamo-cortical loops. Damage at these nodal points will enhance vulnerability to delirium. An important branch of these circuits is the medial forebrain bundle, which carries axons from the ventral tegmentum (dopamine), the locus ceruleus (norepinephrine), and the nucleus raphe (serotonin). Corticostriatal glutamatergic, distributed cholinergic, and GABA-ergic neuronal tracts are also key variables in basal ganglia-thalamo-cortical functioning.
In the striatum, dopamine is required for glutamate to precipitate hypoxic neuroinjury. Dopamine blockade may protect neurons against subsequent hypoxic stress and injury
(22) . Preclinical studies suggest that haloperidol may protect against brain ischemic oxidative stress through its sigma-1 antagonistic butyrophenone effect
(38) .
Clinically, intravenous haloperidol can rapidly improve hippocampal function (e.g., short-term memory) and reverse frontal disinhibition. Dopamine excess may cause agitation and delusions. Acetylcholine insufficiency may lead to disorientation, hallucinations, and memory impairment
(39) . In animals, stress increases mesocorticolimbic dopamine release
(40) . Dopamine, a weak inhibitor of acetylcholine release, in excess may exacerbate the effects of a relative acetylcholine deficiency
(41) .
Psychotic fear in delirium may originate in the central extended amygdala, which normally integrates internal stimuli from the hypothalamus and brainstem and external stimuli from the basolateral amygdala
(42) . In delirium, abnormal signals from the hippocampus, temporal cortex, and medial orbitofrontal cortex may converge on the basolateral amygdala, which subsequently sends disordered inputs to the central extended amygdala. The medial central extended amygdala may then abnormally excite dopamine subpopulations that in turn project to limbic areas and to cognitive regions of the cortex and striatum. This may be why the fear in delirium requires dopamine-blocking neuroleptics rather than benzodiazepines to be managed effectively.
Management and Treatment
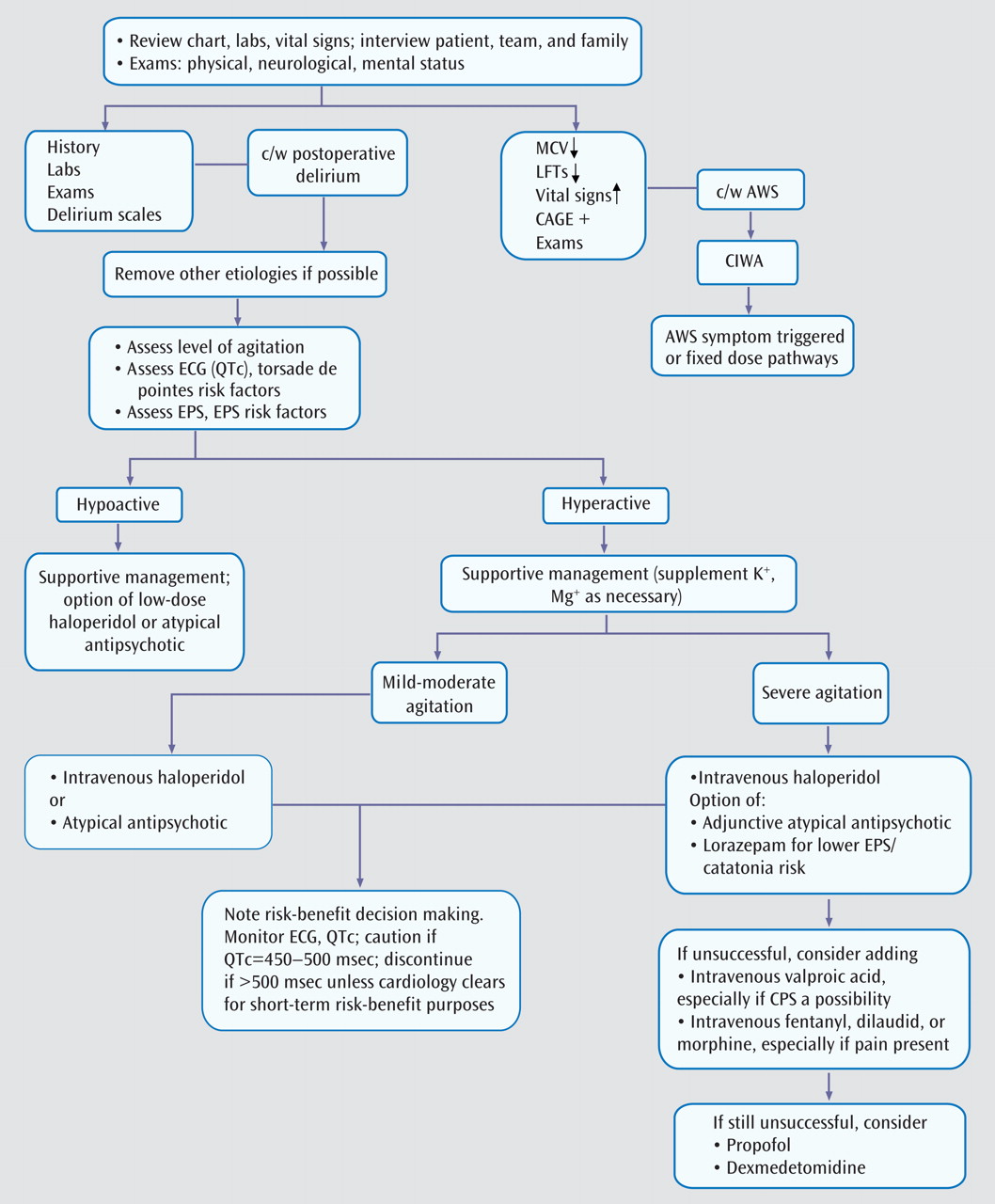
Because postoperative delirium is a diagnosis of exclusion, evaluation of other etiologies is the first step in management and treatment (
Figure 2 ). Eliminating underlying causes can be corrective, and pharmacological agents are among the most common iatrogenic causes of delirium.
Review of current and past medications is essential, since delirium may occur when CNS-active drugs reach toxic levels or when certain drugs and substances associated with physiological dependence are withdrawn abruptly. Drug-drug interactions can further complicate the issue. Management entails discontinuing or reducing the dose of the offending drug(s) when feasible.
If removal of deliriogenic factors fails to lead to improvement and if the patient’s behavior dictates it, the treatment of choice is neuroleptic medication. Haloperidol administered intravenously is the mainstay of the management of delirious agitation, as underscored in APA guidelines
(43,
44), despite the fact that this route of administration has not been approved by the U.S. Food and Drug Administration (FDA) and that haloperidol now carries a black box warning about the potential for lethal ventricular arrhythmias, including torsade de pointes
(45) . At least 28 case reports of QT prolongation and torsade de pointes, some with fatal outcome, have been reported with haloperidol. It may be of anecdotal interest in this regard that with over 90 career years of haloperidol use among the authors, none of us can recall seeing a case of torsade de pointes after initiating intravenous haloperidol, although we have run across other ventricular arrhythmias. When prescribing intravenous haloperidol, one should monitor the ECG and clarify why the risk-benefit ratio favors its use. Haloperidol may be especially effective against agitation because of its strong dopamine D
2 receptor binding affinity, which atypical antipsychotics lack. It has few anticholinergic side effects and few active metabolites. The effects of haloperidol on blood pressure, pulmonary artery pressure, heart rate, and respiration are milder than those of intravenous benzodiazepines and other neuroleptics, and it is less sedating
(43,
44,
46) .
Intravenous administration of haloperidol is preferable to intramuscular administration. Drug absorption may be poor in distal muscles if there is hemodynamic instability. In addition, painful intramuscular injections may exacerbate paranoia. Interpretations of muscle enzyme values may be erroneous with frequent intramuscular injections. Moreover, haloperidol administered intravenously is less likely to produce extrapyramidal symptoms than intramuscular or even oral haloperidol
(47) . Intravenous haloperidol has a mean distribution time of 11 minutes, although in critically ill and elderly patients this interval may be prolonged, and thus tranquilizing effects may take 15–20 minutes. The mean half-life of haloperidol when given intravenously is about 24 hours. Intravenous haloperidol is twice as potent as the oral form
(46) .
We often prescribe 2–2.5 mg i.v. of haloperidol for mild agitation, 5 mg i.v. for moderate agitation, and 7.5–10 mg i.v. for severe agitation. In the elderly, doses should be approximately one-third of what is usually prescribed. The APA guidelines recommend 0.25–0.5 mg every 4 hours as needed for elderly delirious patients
(43,
44) .
Doses can be repeated intravenously every 30 minutes until the patient is calm yet arousable to normal voice. When serious agitation persists, the previous dose can be doubled 30 minutes later; this approach of successive doubling can be repeated. The goal is to ameliorate the agitation rapidly, as partial control can sometimes prolong the delirious state. For reemergent agitation, the last effective dose can be administered. For unremitting agitation, a continuous infusion of 5–10 mg/hour can be instituted, although we have very rarely needed this approach.
Careful, painstaking clinical evaluation should guide any necessary medication adjustments. If sensorium and behavior improve, lower doses of intravenous haloperidol or an orally administered atypical antipsychotic may suffice.
Maximum daily doses have not been established, although over 1000 mg of intravenous haloperidol has been used safely
(48) . Hypotension can occur as a result of a low-volume state; if this occurs, monitored volume replacement can precede administration of any further doses. Haloperidol may lower seizure threshold, although it is one of the safest neuroleptics in this respect. Haloperidol is rarely associated with respiratory depression and is well tolerated by patients with chronic obstructive pulmonary disease. In the ICU setting it can often help reduce fear and agitation associated with ventilator weaning.
Haloperidol infrequently causes ventricular arrhythmia, occasionally in a torsade de pointes configuration, so ECG monitoring is important, especially in susceptible populations
(49,
50) . Drug-induced QTc prolongation is relatively common, whereas QTc greater than 500 msec is uncommon and torsade de pointes is rare. Risk factors include the myocytic drug effect of blocking the delayed rectifier potassium channel (I
Kr ) resulting in decreased K
+ outflux, which is how neuroleptic medications primarily offend. Secondary risk factors amplify a congenital or acquired reduced “repolarization reserve.” Individuals with reduced repolarization reserve cannot compensate for drugs that block I
Kr and threaten depolarization. Secondary risk factors include high doses or rapid administration, inhibition of cytochrome P450 liver enzymes (for example, paroxetine and ketoconazole are inhibitors of haloperidol oxidative metabolism), impaired drug elimination, bradycardia, hypokalemia, hypomagnesemia, left ventricular hypertrophy, heart failure, recent conversion of atrial fibrillation, hypothyroidism, older age, female gender, concomitant use of another I
Kr blocker, undetected ion channel polymorphisms, and congenital long QT syndrome. The vast majority of patients who develop drug-related torsade de pointes have at least one risk factor.
Pharmacodynamic interactions with other QTc-prolonging medications, such as fluoroquinolones, macrolides, class I antiarrhythmics, tricyclic antidepressants, carbamazepine, and methadone, may further contribute to prolonged QTc. Discontinuation or a switch to non-QTc-prolonging medications may be required.
In healthy male subjects, orally administered haloperidol causes a mean increase in QTc of 4.7 msec. Atypical antipsychotics can also increase the QTc, as reported in a Pfizer study for the FDA
(50,
51) : olanzapine causes a mean increase of 6.8 msec, risperidone 11.6 msec, quetiapine 14.5 msec, and ziprasidone 20.3 msec. Aripiprazole may offer a better QTc risk profile and has been used in cases of mild to moderate delirium, as have other atypical antipsychotics
(52,
53) .
In a meta-analysis of two randomized controlled trials—haloperidol versus risperidone and haloperidol versus olanzapine—no evidence of differential effect on symptoms and side effects was noted when low-dose haloperidol was used
(54 –
56) . With haloperidol doses greater than 4.5 mg/day, more extrapyramidal side effects occurred. In neither study was haloperidol given intravenously, nor were possible ethnopsychopharmacological confounders addressed. Nevertheless, the authors of the meta-analysis suggest using atypical antipsychotics instead of orally administered haloperidol in high doses or when there is a high risk of extrapyramidal side effects or cardiotoxicity.
Unfortunately, this is not always a pragmatic suggestion. While atypical antipsychotics given orally or intramuscularly are useful for mild to moderate delirious agitation and sometimes as a supplement for severe and dangerous agitation, the treatment of choice in the latter case, in our view, is intravenous haloperidol in high doses if needed because it offers the safest and quickest way to calm the patient, in whom the risk of insufficiently treated severe agitation often outweighs the risk of side effects. After reviewing the QTc risk factor profile, we make corrections as needed (e.g., supplementation of potassium to 4 mmol/liter and magnesium to 2 meq/liter) before giving intravenous haloperidol. When the QTc increases to 450–500 msec, there is cause for concern and caution; when it exceeds 500 msec, there is an increased risk of torsade de pointes
(49) . We adhere to APA guidelines suggesting that a QTc interval greater than 450 msec or more than 25% over baseline may warrant a cardiology consultation and reduction or discontinuation of the neuroleptic medication. Occasionally, however, when the QTc is greater than 500 msec and a risk-benefit analysis made in concert with the cardiology and medical teams indicates that it is clinically necessary to continue acute, short-term treatment, we continue cautiously with the aid of cardiac telemetry, close monitoring of electrolytes, and elimination of any reversible pharmacokinetic risks and potentially adverse pharmacodynamic interactions.
Intravenous droperidol, which carries a black box warning, can also reduce delirious agitation; however, this agent is a more potent α 1 -adrenergic receptor antagonist and thus is more likely to cause hypotension. It can also be more of a respiratory depressant. Other parenteral neuroleptic drugs occasionally used for treatment of agitation include perphenazine and chlorpromazine, which are effective but unfortunately can cause hypotension and occasionally a decline in cardiac output because of their potent α 1 -adrenergic antagonism. These phenothiazines are relatively anticholinergic, can lower the seizure threshold, and prolong the QTc. However, in low doses they can be both safe and effective.
Adding lorazepam (1–2 mg i.v. every 2–4 hours) to haloperidol in the management of severely agitated patients can be helpful because lorazepam may reduce the extrapyramidal side effects of haloperidol, especially akathisia
(57,
58) . Lorazepam given intravenously has a half-life of about 12–14 hours; therefore, its sedating effect may accumulate, and tapering before ventilator weaning may be needed. Nevertheless, lorazepam is one of the least likely benzodiazepines to cause respiratory depression. Along with oxazepam and temazepam, it is safest for patients with hepatic insufficiency because it does not require oxidative metabolism.
The addition of intravenous valproate may be considered to manage severe agitation when conventional therapy is inadequate or when problematic side effects emerge
(59) . Fentanyl or hydromorphone can be helpful for severely agitated patients on mechanical ventilation. If these medications are not sufficiently calming, sedation with ventilator support can be achieved with propofol or dexmedetomidine. Propofol is deeply sedating and is short acting. It has a titratable progression from anxiolysis to sedation to hypnosis to anesthesia
(60) . However, when used over a prolonged period (≥2 weeks), tachyphylaxis can emerge, making weaning from propofol difficult
(61) . In these situations, we have successfully used dexmedetomidine, a very selective α
2 -adrenergic receptor agonist, with caution regarding low-output cardiac side effects. Dexmedetomidine warrants further study in postoperative delirium
(62) .
Pain can exacerbate agitation in postoperative delirium. Morphine, hydromorphone, or fentanyl can be used. Respiratory depression and hypotension are potential side effects. In patients being managed with fentanyl, it is occasionally difficult to achieve steady-state tranquilization because of this agent’s short-acting nature. Meperidine is to be avoided because of deliriogenesis related to its normeperidine metabolite. Patient-controlled analgesia and patient-controlled epidural analgesia using an opioid and a local anesthetic are important strategies for controlling pain after surgery. In one randomized study of elderly patients who had undergone major abdominal surgery, both of these methods were effective, but patient-controlled epidural analgesia was actually associated with better pain, mental status, and bowel outcomes over the 5-day postoperative study period
(63) .
Specific nonpharmacological management strategies for delirium have been advocated
(5,
10) (see
Table 1 ). At present, evidence on delirium prevention is sparse, but more research is on the way
(64) .