Mood symptoms and syndromes are common during and after pregnancy and are potentially harmful to both mother and infant. While genetic factors clearly influence mood disorders in general, evidence for a genetic role specific to postpartum symptoms is less well-established, as this area has been little explored. Postpartum depression occurs in up to 10%–20% of mothers in the year following delivery
(1,
2) . The risk for postpartum depression is increased in women with a history of major depression
(3,
4) and in women with a history of postpartum depression following previous pregnancies
(5,
6) . Our group found that approximately 20% of women with major depression report depressive symptoms in the month following delivery
(7) . In women with bipolar disorder, postpartum mood episodes including both depression and mania have been reported at rates as high as 25%–50%
(8) ; the specific risk of postpartum psychosis, a syndrome resembling mania with psychotic features, is 20%–30%
(9 –
11), although it remains unclear if prophylactic treatment lowers this rate. We have shown that approximately 20% of women with bipolar disorder report significant mood symptoms within a month of childbirth and that approximately 50% experience significant symptoms either during pregnancy or postpartum
(7) .
A genetic basis for postpartum mood syndromes is suggested by several studies. Family studies of postpartum psychosis have supported a genetic susceptibility to a postpartum trigger in bipolar disorder, as well as an overlap in genetic factors predisposing to postpartum psychosis and bipolar disorder
(12,
13) . Dean et al.
(14) found a higher risk of postpartum mood illness in relatives of probands with postpartum psychosis. Forty et al.
(15) showed that the trait of postpartum depression exhibited familiality in pedigrees with recurrent major depression. We have reported familial aggregation of postpartum depressive symptoms in families with recurrent early onset major depression and bipolar disorder
(16,
17) . There has been one genome-wide linkage study related to postpartum mood symptoms, narrowly focused on postpartum psychosis
(18) . To date there has been no linkage study of postpartum mood symptoms nor one focused on depressive symptoms in this setting.
We therefore undertook a genome-wide linkage scan of postpartum mood symptoms in pedigrees with major depression or bipolar disorder. We made use of large genotyped family sets available for both disorders through two collaboratives. We further had the benefit of data from genome-wide association studies conducted by the collaborative groups, with which we fine-mapped our linkage findings.
Results
Linkage
The final linkage dataset included genotypes from 1,210 informative women, 757 from GenRED and 453 from NIMH-BP. Complete information about the timing of mood symptoms in relation to childbirth was not available for 84 subjects. Of the remaining 1,126 subjects, 258 (22.9%) had postpartum mood symptoms. The mood disorder diagnoses present in this sample included recurrent major depression (62.6%), bipolar I disorder (26.9%), single-episode major depression (3.9%), bipolar II disorder (3.4%), schizoaffective disorder-bipolar type (1.3%), and other mood disorders (1.9%). There were no differences between the NIMH-BP and GenRED samples in mean age at onset of mood disorder (20.5 and 20.0 years, respectively; t=0.91, p=0.3633), mean age at interview (44.3 and 45.0 years; t= –1.01, p=0.3129) or rate of postpartum mood symptoms (21.4% and 23.6%; χ 2 =0.70, p=0.4019).
Table 1 shows the numbers and types of informative affected relative pairs for all women in the baseline group and for the subset with postpartum mood symptoms. There were 961 informative affected relative pairs for the baseline linkage analysis and 63 for the analysis among the subset with postpartum mood symptoms.
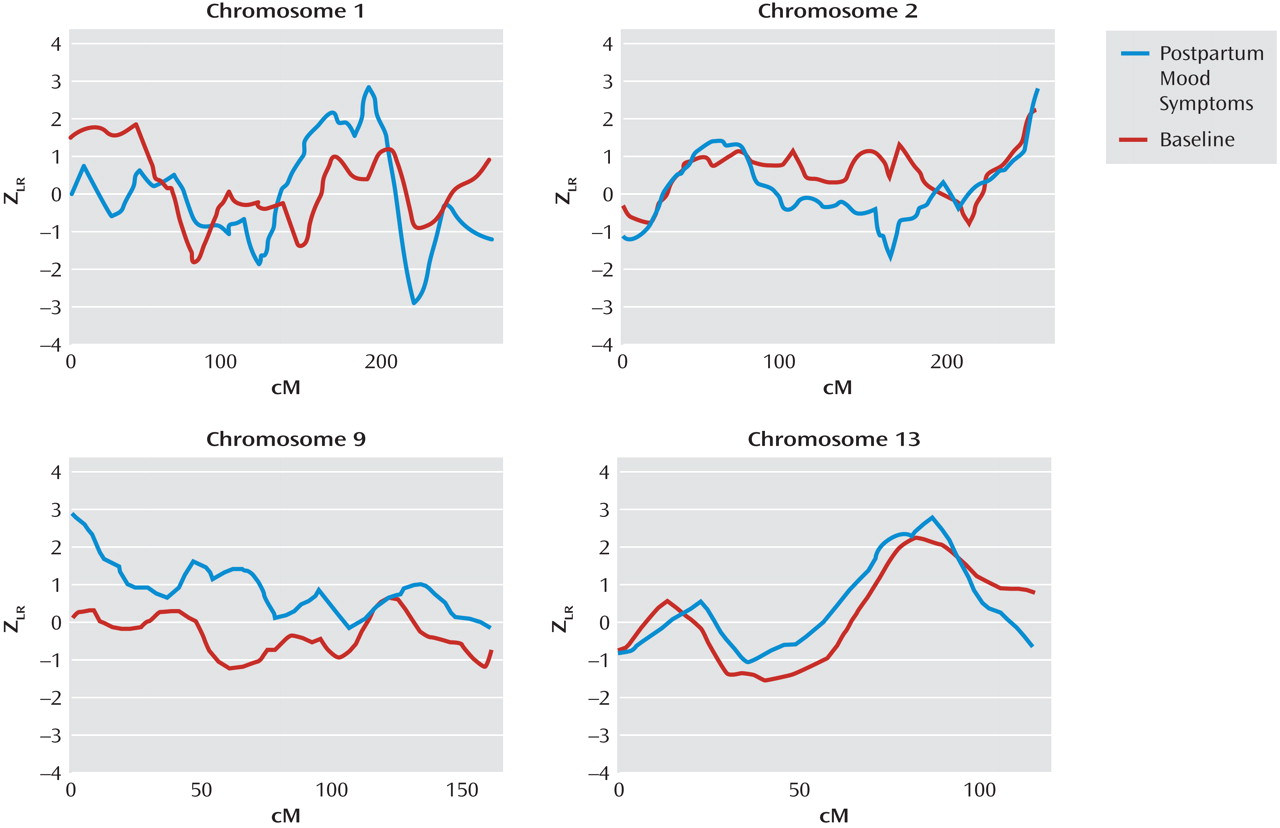
Figure 1 illustrates the results for our top four chromosomal regions, based on the maximum Z
LR score in the linkage analysis for the subset with postpartum mood symptoms. Complete genome-wide results are provided in Supplementary
Figure 1 that accompanies the online version of this article. A maximum Z
LR of 2.93 was observed in the postpartum mood symptom group for marker D1S1660 at 189.3 cM on chromosome 1. Permutation testing showed this result was chromosome-wide significant (p=0.02), although it did not reach genome-wide significance (p=0.35). At this location, the Z
LR in the baseline group was only 0.32, so the increase from baseline was 2.61 in the postpartum mood symptom group (empirical p=0.004). The 2-Z
LR support region around the linkage peak included the chromosomal region 1q21.3-q32.1, spanning 60 cM and containing seven markers genotyped across all samples. The mean information content value in the postpartum mood symptoms analysis was 0.715 genome-wide and 0.783 in the 2-Z
LR region. Other linkage peaks (maximum Z
LR >2.0) in the postpartum mood symptoms analysis were seen on chromosomes 2q37.1-q37.3, 9p24.3-p22.3, and 13q21.33-q33.1 (
Figure 1 ). However, of these regions, only a 29-cM region on 9p24.3-p22.3 showed a significant difference between the postpartum mood symptom group and the baseline group (change in Z
LR =2.77, empirical p=0.001).
Association
Encouraged by the linkage specific to postpartum mood symptoms on chromosomes 1q21.3-q32.1 and 9p24.3-p22.3, we sought to follow-up these findings in a densely genotyped SNP association study. Of the 759 women who met our baseline criteria and had genotype data available for this analysis, 457 were from GenRED and 302 from NIMH-BP. The distribution of mood disorder diagnoses was as follows: recurrent major depression, 58.8%; bipolar I disorder, 38.7%; single-episode major depression, 1.4%; and schizoaffective disorder-bipolar type, 1.1%. The NIMH-BP subjects were slightly older at interview than the GenRED subjects (45.2 versus 43.6 years, respectively; t=1.98, p=0.0483). There was no difference in the proportion of women with postpartum mood symptoms between the NIMH-BP (25.2%) and GenRED (24.3%) samples (χ 2 =0.08, p=0.7838). Overall, 187 women (24.6%) met our criteria for postpartum mood symptoms.
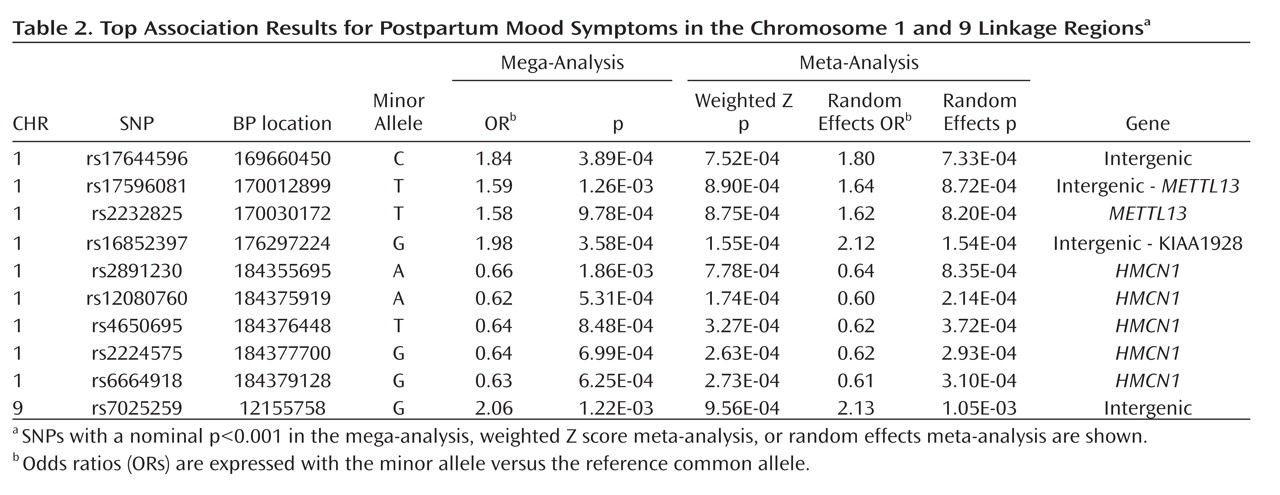
Our top case-only meta-analysis and mega-analysis association results are summarized in
Table 2, which lists all results with p<0.001 using any method. All results from our two linkage regions are presented in Supplementary
Figure 2 . Our best finding was on chromosome 1 for SNP rs16852397, with a mega-analytic odds ratio of 1.98 (p=3.58×10
–4 ). This SNP was also nominally associated with postpartum mood symptoms in the meta-analytic methods controlling for population stratification. rs16852397 is intergenic but is located in a spliced EST. Our best association signal on chromosome 9 was for the intergenic SNP rs7025259 (meta-analytic p=9.56×10
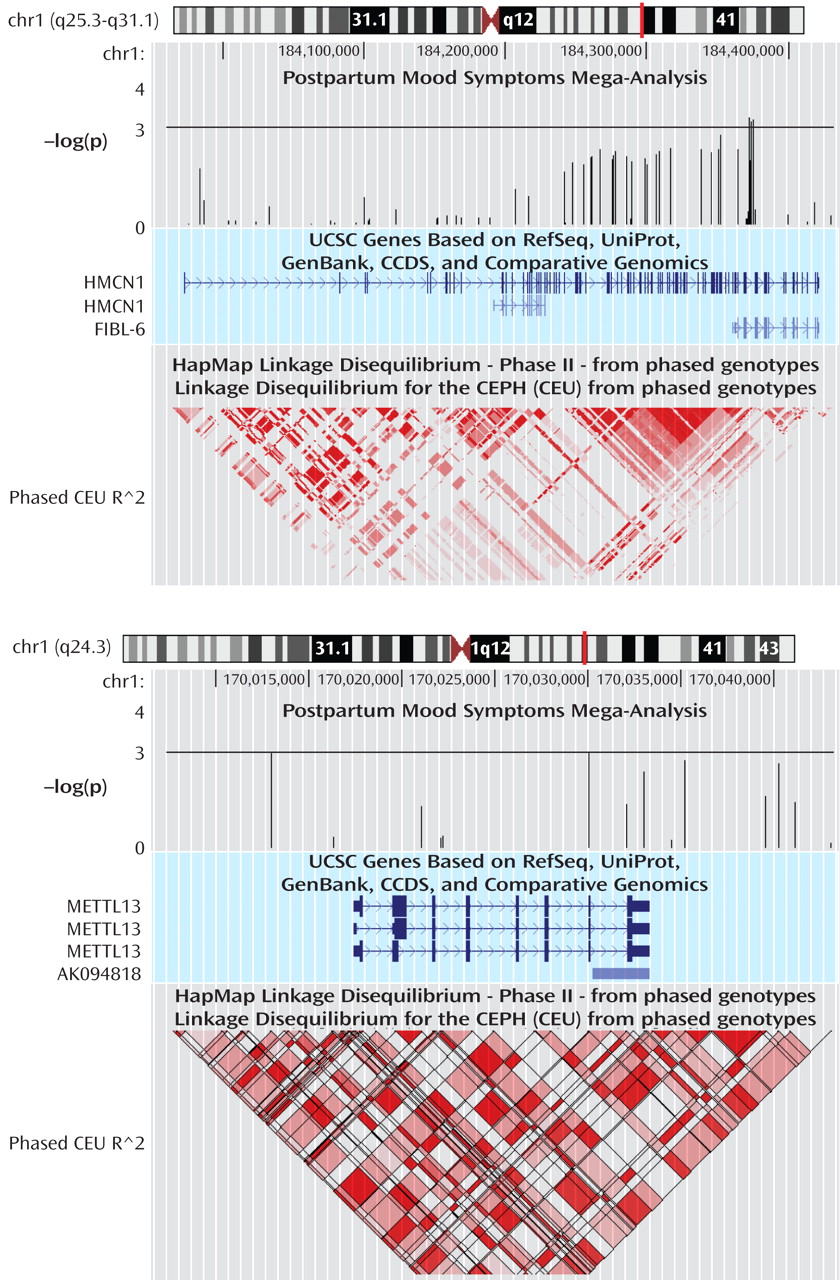
–4 ). Two genes that were implicated among our top findings are
HMCN1 (Hemicentin 1) and
METTL13 (Methyltransferase like 13) on chromosome 1 (
Figure 2 ). None of these findings were significant after accounting for the multiple SNPs tested across the regions. For our top results, there was no evidence of between-sample heterogeneity (Q-statistic p>0.05). We tested whether SNPs in our two top genes of interest showed evidence of interaction in relation to postpartum mood symptoms. The strongest interaction between two SNPs across these genes was nominally significant (p=0.0027). This does not hold up to multiple testing given that 924 comparisons were made between 66 SNPs in
HMCN1 and 14 SNPs in
METTL13 .
Discussion
To our knowledge, this is the first study to examine the genetic etiology of postpartum mood symptoms using a systematic, genome-wide approach. We observed genome-wide suggestive linkage signals on 1q21.3-q32.1 and 9p24.3-p22.3 that may be specific to postpartum mood symptoms. We followed up these 2-Z LR regions surrounding the linkage peaks on chromosomes 1 and 9 using a SNP association study. Our best association signal on chromosome 1 was for the intergenic SNP rs16852397 and on chromosome 9 was for the intergenic SNP rs7025259. We also found modest evidence of association for SNPs on chromosome 1 in the genes HMCN1 and METTL13 with postpartum mood symptoms.
While ours is the first genome-wide linkage study of postpartum mood symptoms, one previous study examined linkage in postpartum psychosis
(18) . Using the Wellcome Trust UK-Irish Bipolar Sib-pair sample and analyzing only families in which at least one woman had an episode of postpartum psychosis, Jones et al. reported genome-wide significant linkage on chromosome 16p13 and suggestive linkage on 8q24. Neither of these regions reached nominal significance in our study. It is worth emphasizing that in our study less than 30% of the subjects were diagnosed with bipolar I disorder, and that our definition of postpartum mood symptoms was much broader than that used by Jones et al. Our best linkage peak on chromosome 1 was previously reported to show modest evidence of linkage in Ashkenazi Jewish bipolar disorder families
(36) . This finding is consistent with our observation of a modest linkage signal in this region in our baseline group, and an enhanced signal in the group with postpartum mood symptoms. No evidence of linkage with mood disorders has been previously reported in our region on chromosome 9p.
Motivated by the hypothesis that peripartum mood syndromes may have a unique genetic etiology, several studies have examined candidate genes for association with peripartum psychosis. Polymorphisms in the serotonin receptor gene
HTR2C and the serotonin transporter
SLC6A4 have been found to be associated with peripartum psychosis
(37) . However, no associations have been reported with polymorphisms in the genes
ESR1, HTR2A, NR3C1, and
TNFA (37 –
41) .
HMCN1 and
METTL13 have not been previously examined for association with peripartum mood syndromes.
HMCN1 is 456 kb long and codes for an extracellular matrix protein with several functions, including transmembrane receptor activity and calcium ion binding. The gene contains four experimentally determined estrogen receptor binding sites
(42), which might be relevant for a postpartum phenotype.
HMCN1 is particularly highly expressed in the hippocampus
(43), a brain region likely to be involved in depression
(44), and shown to be altered in rats by a postpartum drop in estrogen levels
(45) . Twenty-seven SNPs in
HMCN1 were at least nominally significant (p<0.05), suggesting our results are not due to genotyping error. Our genotyped SNPs in
HMCN1 passing quality control captured 83% of the common variation in the gene (MAF≥0.01, r
2 ≥0.8). The gene
METTL13 is putatively involved with methyltransferase activity and is 16 kb in length. Interestingly, DNA methyltransferases have been shown to play a role in estrogen receptor-induced gene transcription
(46) . Six of the available SNPs passing quality control in
METTL13 were at least nominally significant. The genotyped SNPs captured 88% of the common variation in this gene.
Our results should be viewed in light of several limitations. First, these samples were originally ascertained for other purposes. While the clinical data were collected using a rigorous and well validated instrument, the interview did not contain all possible information about peripartum mood symptoms. Second, we combined samples from different sources for this study, potentially introducing heterogeneity. We combined these samples to create one of the largest datasets available to examine the genetic etiology of postpartum mood symptoms. We felt this was appropriate as the samples used similar ascertainment, assessment, and genotyping methods. Third, prospective data might provide greater clarity for assessing the timing of the onset of symptoms relative to parturition. Fourth, we corrected the meta-analysis for population stratification but not the mega-analysis, as it can lead to overconservative estimates of effect sizes. This may have contributed to the slight differences observed between the results from these two approaches. Finally, even though we combined samples to create a larger dataset, we still had limited power to detect loci of modest effect.
In conclusion, we performed the first genome-wide linkage analysis of postpartum mood symptoms using a large sample with detailed clinical information. We followed up our best linkage peaks in an association study with densely genotyped SNPs. Our results suggest there may be genetic variation contributing to susceptibility to postpartum mood symptoms in the 1q21.3-q32.1 and 9p24.3-p22.3 regions. Specifically, the genes HMCN1 and METTL13 may contain polymorphisms that confer susceptibility to postpartum mood symptoms. As both the linkage and association results presented here are novel, future studies replicating these findings are warranted.