The popular expression "What does not kill you makes you stronger" points to the fact that some people respond resiliently to trauma. This statement may be true for highly resilient people. However, for those who are vulnerable, a more appropriate statement might be "What does not kill you can make you ill." Such vulnerability is common. Approximately one-tenth of those who survive life-threatening events will develop mental health disorders such as posttraumatic stress disorder (PTSD) or depression or both (
1,
2). One of the goals of modern psychiatry is to identify vulnerable individuals and intervene to prevent the development of these disorders by bolstering resiliency. The factors that contribute to resiliency encompass both biological and psychological aspects of the individual as well as the pre- and posttrauma environment (
3). It has also been suggested that resiliency is a product of early stress—that is, that resiliency is an adaptive response that maintains homeostasis under stressful circumstances (
4). However, this response is true only for some individuals; for others, traumatic stress can increase vulnerability.
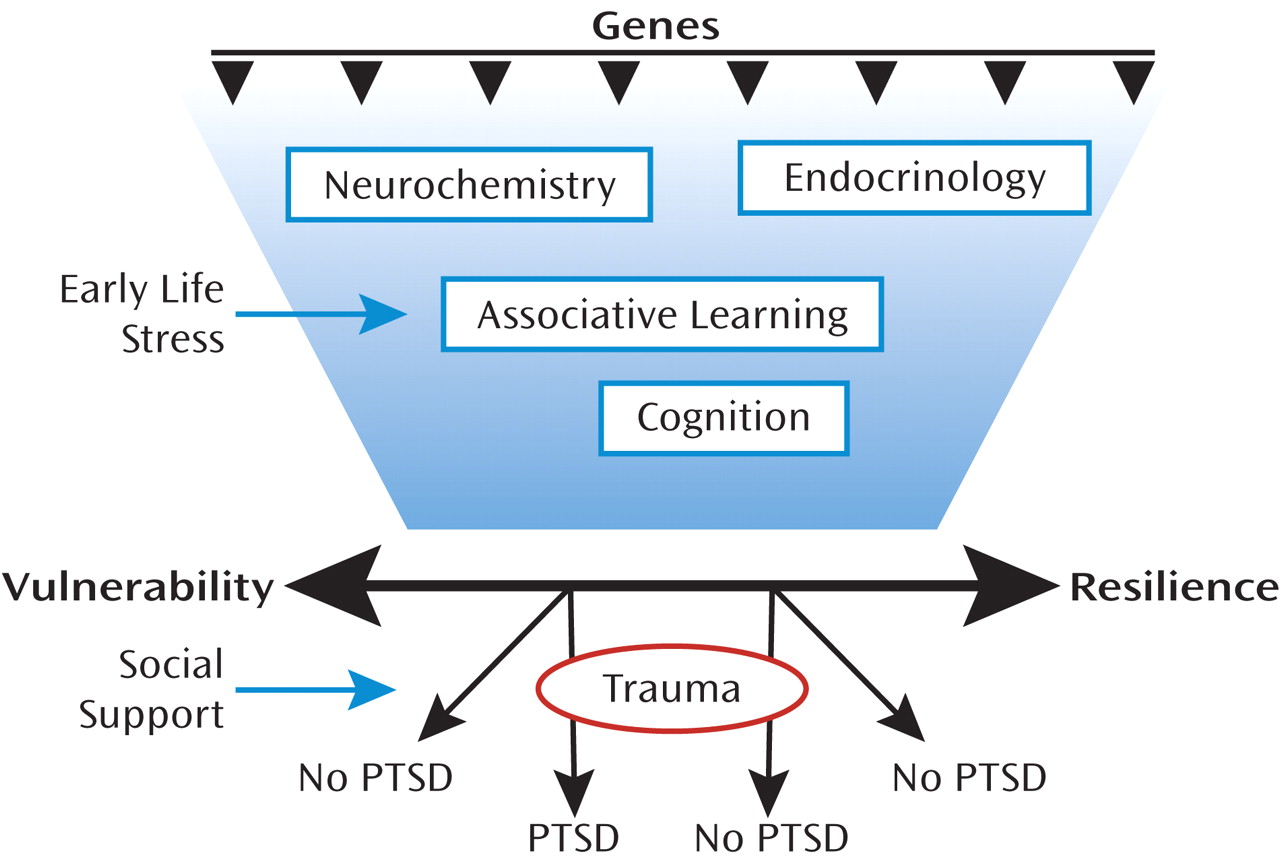
Resiliency results from a combination of both biological factors (which are heritable) and environmental factors to which the individual is exposed (
Figure 1). The environmental factors that promote resiliency, including social support after trauma, have been the focus of treatment for PTSD. The biological factors may be based on heritable genetic profiles, which may code for neurochemicals and neural mechanisms that promote resiliency. Recent studies have shown that specific gene alleles are associated with resilience, such that even severe levels of child abuse do not result in severe psychopathology (
5,
6). The genetic profile may also code for associative learning mechanisms, such as fear conditioning, to enhance fear responses or to enhance fear extinction, which promotes suppression of fear responses to previously fearful stimuli (
3).
Vulnerability to the development of PTSD after trauma exposure may be associated with an exaggerated fear response or an inability to control fear responses, which could either be a risk factor for the disorder (
7) or an acquired trait of the illness (
8). The DSM-IV (
9) diagnosis of PTSD requires exposure to a traumatic event and a cluster of symptoms associated with that event (e.g., psychological and physiological reactions to trauma reminders and avoidance of such reminders). Consequently, several theorists (see reference
10, for example) have proposed that conditioning processes are involved in the etiology and maintenance of PTSD. Especially pertinent to this view is the idea that through the processes of Pavlovian conditioning, a neutral (conditioned) stimulus that occurs in temporal contiguity with an aversive (unconditioned) stimulus that innately elicits pain and fear acquires the ability to elicit a fear response in the absence of the unconditioned stimulus. Thus, neutral stimuli (the conditioned stimuli) present at the time of the trauma (the unconditioned stimulus) acquire the ability to elicit a conditioned fear reaction that can be triggered when the person subsequently encounters these or similar stimuli during the course of normal life. Consistent with this hypothesis, emotional and physiological reactivity to stimuli resembling the original traumatic event even years after the event's occurrence is a prominent characteristic of PTSD and has been reliably replicated in the laboratory (see references
11–
13, for example). While PTSD is a complex disorder that includes the dysregulation of other emotions besides fear, such as anger or guilt, and is highly comorbid with depression, the study of fear lends itself best to translational approaches (
14–
16). In this review we first focus on the neural circuits that are involved in inhibition of fear responses and then discuss recent genetic findings in the area. We conclude with a discussion of how these results may be combined in a neurogenetic model that incorporates risk and resilience to trauma-related disorders and indicates prevention and treatment targets in the time course of development of the disorder.
Fear Inhibition as an Intermediate Phenotype
Fear inhibition involves learning of safety signals—that is, the ability to discriminate between danger and safety cues and to suppress fear responses in the presence of safety cues. In the laboratory fear inhibition can be measured by first using a fear conditioning paradigm for fear acquisition, which is then followed by the training of fear inhibition. Fear conditioning is based on a simple Pavlovian conditioning model, in which a neutral conditioned stimulus (CS; for example, a light) is paired with an aversive unconditioned stimulus (US; for example, an electric shock). After a number of pairings, the association is formed so that the CS alone elicits the conditioned response (CR; for example, a fear response). This basic model is used in animal as well as human research to investigate mechanisms of fear acquisition.
Two major laboratory models have been used for behavioral testing of fear inhibition in animals and humans: extinction and conditioned inhibition. While fear acquisition refers to learning that something is dangerous, extinction is a mechanism by which an individual learns that something that previously elicited fear is no longer dangerous—that is, that it is safe. In fear extinction paradigms, a stimulus that was previously paired with an aversive stimulus (the CS+) is then repeatedly presented without the US, so that it no longer elicits a fear response (
17,
18). In a basic conditioned inhibition paradigm, the above CS+ pairing is intermingled at the time of training with a separate stimulus (CS–). In other words, the CS– does not co-occur with an aversive stimulus and thus represents safety or inhibition of fear. In another standard conditioned inhibition paradigm, one cue is paired with the aversive stimulus when presented alone (CS+, also referred to as A+) but not when presented in compound with a second cue (CS–, represented as AX–, indicating that the combination of A and X is not reinforced). In this model X should become a safety signal because it signals the absence of the aversive stimulus (
19).
In humans, two physiological responses have been used as behavioral outcome measures for fear conditioning: acoustic startle response and skin conductance response. The acoustic startle response is characterized by an integrative reflex contraction of the skeletal musculature in response to a strong stimulus. It provides an excellent model to study emotional processing since the amygdala is directly connected with the startle circuit (
16,
20–
22). Fear-potentiated startle is the relative increase in the acoustic startle response elicited in the presence of a conditioned stimulus (CS+) that was previously paired with an aversive stimulus (US). The skin conductance response is an index of sympathetic nervous system activity that is frequently used in measuring fear acquisition and extinction in tandem with brain imaging studies using positron emission tomography (PET) or functional MRI (fMRI) (
8,
23–
26).
Unfortunately, traditional conditioned inhibition paradigms have a number of confounding issues, such as second-order conditioning, external inhibition, and configural learning, that make it difficult to discretely separate excitatory fear learning from inhibition of fear in neural circuits. Myers and Davis (
27) developed an animal model using a
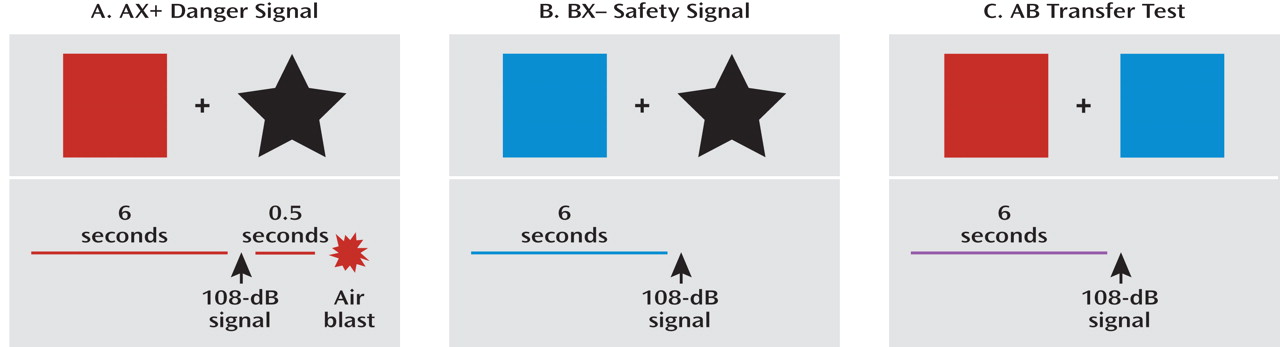
conditional discrimination procedure that allows for the independent evaluation of excitation and inhibition of fear conditioning. In collaboration with this group, we developed a conditioned inhibition paradigm for use in humans (
28) that contains a danger signal (AX+), a safety signal (BX–), and a safety transfer test (combination of A and B, where B reduces fear to A) (
Figure 2). The procedure, referred to as a conditional discrimination (abbreviated as AX+/BX–), is based on a paradigm used in earlier learning theory experiments (
29,
30). In this experiment, the response to stimulus X is conditional on the presence of either A or B. The A stimulus elicits fear potentiation of startle with training as the subject learns that A and X presented together predicts the US. Stimulus B elicits reduced startle compared to A (i.e., becomes inhibitory) in that B presented with X predicts safety from the US. The presentation of AB results in a reduced startle response compared to the response to A presented with a neutral stimulus because B has become inhibitory.
We translated this paradigm to use in clinical settings and have now demonstrated conditioned inhibition in healthy individuals (
28) and in combat veterans with low levels of current PTSD symptoms (
31). On the other hand, study subjects with high levels of PTSD symptoms were unable to reduce startle to AB trials (i.e., were unable to transfer fear inhibition). We have also replicated these findings in a sample of veterans with PTSD from the University Hospital Dubrava in Zagreb, Croatia (
32), and in a civilian population in Atlanta with high levels of urban trauma (
32a). Together these data suggest that PTSD is at least in part a disorder in which inhibition of fear is deficient, even when learned fear in the laboratory is separate from the index trauma(s). An alternative explanation is that fear excitation to the A stimulus in the AB compound is so exaggerated in PTSD that it overwhelms the inhibition from B. This would be consistent with our findings in Vietnam veterans, in that those with the most severe symptoms also had significantly more fear potentiation to the AX+ cue compared to healthy comparison subjects (
31). However, our replication samples of combat-related PTSD in Croatia and civilian PTSD in Atlanta did not have increased potentiation to AX+; the group differences were limited to AB and BX– trials, which suggests a selective deficit in fear inhibition.
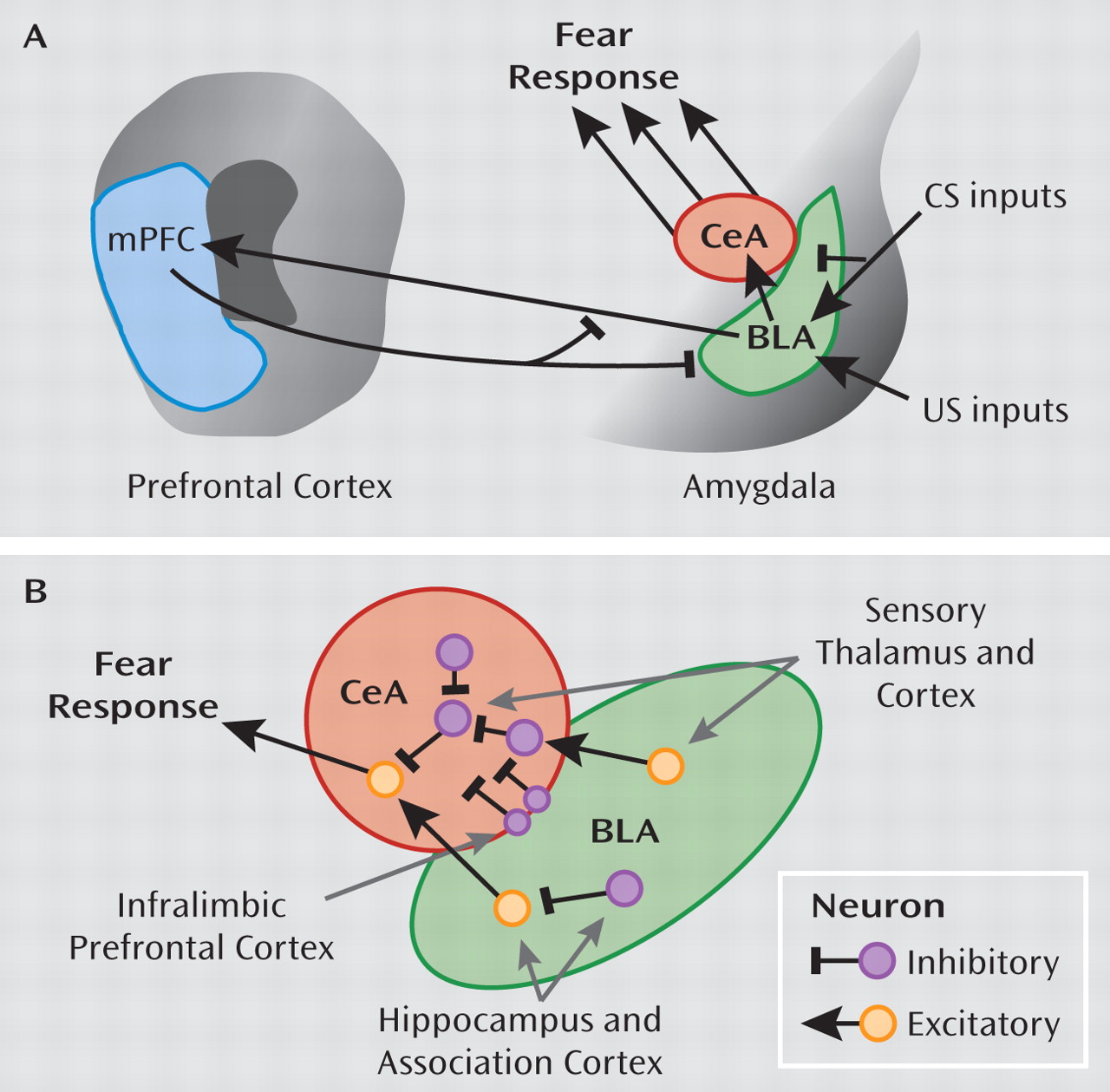
Both extinction tests and conditioned inhibition focus on active suppression of fear responses through learned safety signals; while fear itself may involve only subcortical areas of the brain located primarily in the limbic circuitry, safety signals may require a cognitive, cortical component (
26,
33). This premise is supported by data from our laboratory showing that awareness of the association between the CS and the US is necessary for inhibiting fear responses on the AX+/BX– paradigm (
34). Furthermore, a recent study by Weike and colleagues (
33) examined the temporal domain of fear conditioning with a danger and safety signal and found that safety signal processing was slower than danger processing. The authors argued that top-down cognitive processes are involved in responses to safety signals, which accounts for the latency in response.
A recent meta-analysis of 15 studies using fear conditioning found that patients with anxiety disorders showed greater levels of fear responses compared to healthy comparison subjects (
35). These data suggest that the fear response is overactive or that the inhibition of fear is deficient in PTSD, which has led researchers to use fear conditioning models to examine some of the core PTSD symptoms. One study (
36) used a fear-potentiated startle paradigm with veterans diagnosed with PTSD and found equivalent levels of fear potentiation to the danger signal in the PTSD and comparison groups. However, participants with PTSD also potentiated to the safety cue, whereas the comparison subjects did not. Our recent data also show that increased fear responses to safety cues are related to the severity of current PTSD symptoms (
31). A recent study of patients with panic disorder (
37) found that these patients also had increased fear-potentiated startle responses to the safety cue; this finding may have been related to the patients' increased expectancy of the US during the safety cue. In that study the impaired discrimination between danger and safety appeared to involve both cognitive and physiological deficits. In our study of veterans with PTSD (
31), we observed a dissociation between participants' cognitive awareness (they reported that they did not expect to receive an air blast US during the CS– trial) and startle response, which was potentiated in response to the nonreinforced stimulus. On the other hand, a study by Orr and colleagues (
38) that used skin conductance to examine fear conditioning in PTSD patients found that patients discriminated between the danger and safety cues better than did comparison subjects. In another study (
39), similarly enhanced conditionability in PTSD patients was found when trauma-related cues were used as the US in fear conditioning; the enhanced fear conditioning was also related to slower extinction. Deficient fear extinction in PTSD has been found in several studies that used skin conductance as the physiological measure (
26,
39,
40). A recent study of combat-exposed Vietnam veterans and their non-combat-exposed twins (
41) found that combat-exposed veterans with PTSD did not have impaired extinction learning but rather had less extinction retention on the day after acquisition and extinction compared to exposed veterans without PTSD. Furthermore, impaired retention of extinction appeared to be an acquired trait related to the disorder since the twins of the veterans with PTSD did not show the same impairment.
While some data with combat veterans suggest that impaired fear inhibition may be an acquired trait (
41) associated with current symptom severity (
31), other studies have reported that heightened fear responses and decreased inhibition of fear may be predictors of the disorder. A prospective study of police academy cadets (
42) found that greater skin conductance responses to threatening stimuli and slower habituation prior to trauma exposure were predictive of PTSD symptom severity after trauma exposure. A similar prospective study with firefighters (
7) found that reduced extinction of fear-conditioned responses before the index trauma explained almost one-third of the variance in PTSD symptom severity in later traumatized individuals. It is possible that a decreased ability to inhibit fear is a risk factor for developing PTSD and contributes to the maintenance of the disorder, while decreased extinction retention is a state resulting from the disorder—given that these fear-inhibition phenotypes may have different neural underpinnings, this would explain the above studies.
For the purposes of this review, we will not define fear inhibition as either a vulnerability or an acquired trait of the disorder; more research is needed before such a determination can be made. However, the issue of whether it is a predisposition or a part of the PTSD syndrome itself does not dismiss the utility of impaired fear inhibition as a phenotype. With the development of new techniques for studying fear acquisition and fear inhibition in animal and human subjects, we can begin to understand how the neurobiology of fear is altered in PTSD. Below we review the animal and human data for some of the primary structures involved in fear conditioning and fear inhibition.
Molecular and Genetic Mechanisms of Fear Inhibition
On a molecular level, fear conditioning involves new learning mediated by synaptic plasticity in the amygdala. Both associative fear conditioning and extinction of conditioned fear, a learning process by which a CS is no longer associated with the US, are dependent on activation of glutamate
N-methyl-
D-aspartate (NMDA) receptors. Administration of NMDA receptor antagonists either systemically (
91,
92) or by direct infusion into the basolateral nucleus (
93,
94) prior to extinction training blocks the extinction of fear memories. In addition, blockade of NMDA receptors after extinction training also impairs extinction, which suggests that NMDA receptors participate in the consolidation of extinction memories (
95). In addition to these data, there is evidence that voltage-gated calcium channels are involved in mediating calcium-dependent synaptic plasticity, which may underlie extinction (
96,
97). Additionally, a significant amount of data implicate brain-derived neurotrophic factor (BDNF) in the plasticity underlying fear and extinction learning through its TrkB receptor (
74,
98).
There are also substantial data indicating that regulation of the inhibitory neurotransmitter γ-aminobutyric acid (GABA) is altered differentially in the acquisition of fear versus extinction. Injection of an inverse agonist, FG7142, which blocks GABA function, was shown to block the context-specific effects of extinction learning (
99). Gephyrin, a scaffolding protein involved in GABA insertion into the surface membrane, is decreased at the protein and mRNA level in the amygdala following fear learning and is increased in the amygdala with extinction learning (
100,
101). These data are consistent with enhanced amygdala excitability with fear learning and an increase in amygdala inhibitory tone following extinction. One study (
102) demonstrated that blockade of GABA insertion within the amygdala impairs extinction of conditioned fear. Overall these data suggest that modulation of GABA-ergic microcircuitry within the central and basolateral nuclei is critically involved in the regulation of fear and its inhibition with extinction learning.
These molecular data suggest that expression of genes associated with neural plasticity (e.g., BDNF and glutamate receptors), neural inhibition (e.g., GABA receptors and cannabinoid receptors), and stress responsiveness (e.g., glucocorticoid receptors and corticotropin receptor) may be associated with the learning of extinction or impaired fear inhibition. Abnormal fear acquisition or inhibition, as described above, appears to be associated with PTSD, either as vulnerability factors preceding trauma exposure or as a consequence of trauma-related fear conditioning.
To date, much of the research on the genetic basis for PTSD has been gathered via twin studies. Data from these studies indicate that heritability accounts for 30%–40% of the variance in risk for PTSD (
103–
105). Despite known genetic contributions to risk for PTSD, there have been no linkage studies and only a handful of candidate gene association studies examining genetic main effects to date. In general, these studies have revealed that there are complex interactions between genetic and environmental factors and that many of the identified genetic polymorphisms are in the regulatory promoter regions and not necessarily in the coding regions (
106,
107).
Several recent reviews have examined the genetics of PTSD (
108,
109,
110), so we will not go into detail on this issue here. No genes have yet been reported that appear to have large main effects across the expected several replications in association with PTSD. Among the more replicated findings to date showing an effect are genes encoding the dopamine receptor 2 (
111,
112) and the serotonin transporter (
113,
114). However, these findings have not been uniformly replicated (
115). As for the genes described above, although two studies have reported no associations with BDNF (
116), there have been no reported studies, to our knowledge, of glutamatergic plasticity-related genes, and only one study has sought genetic links between GABA and PTSD (
117). In sum, little is known about the genetic mechanisms of PTSD, including the potential genetic role of the glutamatergic and GABA-ergic systems.
Gene-by-Environment Interactions in PTSD
Although a small but growing body of psychiatric research has identified gene-by-environment interactions predicting other mental disorders or associated symptoms, to date only a handful of published studies have presented data on a gene-by-environment interaction predicting PTSD. The first focused on the serotonin promoter length polymorphism (5-HTTLPR), which was originally described as interacting with level of prior stress to predict adult depression (
118). Kilpatrick and colleagues (
119) identified a gene-by-environment interaction predictive of PTSD in an analysis of individuals exposed to hurricanes in south Florida. The study also classified PTSD patients according to the degree of social support available following trauma exposure. As part of the same hurricane study, Koenen and colleagues (
120) reported that the "s" allele of the 5-HTTLPR polymorphism was associated with a lower risk of PTSD in low-risk environments (low crime and unemployment rates) but a higher risk of PTSD in high-risk environments. These results suggest that social environment modifies the effect of 5-HTTLPR genotype on PTSD risk.
The other primary series of studies exploring gene-by-environment effects examined FKBP5, a protein that modulates the glucocorticoid receptor (GR). FKBP5 is a co-chaperone that regulates GR sensitivity (
121). Its binding to GR decreases nuclear translocation and decreases GR sensitivity. FKBP5 mRNA and protein expression are induced by GR activation, providing an ultrashort feedback loop for GR sensitivity. Polymorphisms in FKBP5 have been shown to associate with differential up-regulation of FKBP5 following GR activation and with differences in GR sensitivity and stress hormone system regulation. Because of the known role of abnormal GR sensitivity in PTSD (
122,
123),
FKBP5 appeared to be a good candidate gene that may underlie this component of PTSD and perhaps risk for PTSD. Additionally,
FKBP5 polymorphisms had been reported to associate with peritraumatic dissociation (a known risk factor for PTSD) in medically injured children (
124). Finally, FKBP5 blood mRNA levels have been found to differentially associate with PTSD in two separate studies (
125,
126). In the largest candidate gene study of PTSD to date (
5), our group showed a gene-by-environment effect of the polymorphisms in
FKBP5 with a history of childhood maltreatment to predict level of adult PTSD symptoms in a traumatized civilian sample. Notably, no main effects were found for the
FKBP5 genotype directly associating with PTSD symptoms, nor was there an effect of the gene interacting with adult trauma levels. These data suggest that the interaction of trauma, perhaps during a developmentally critical period, with the hypothalamic-pituitary-adrenal (HPA) axis stress-related genes alters amygdala regulation of fear and its inhibition later in life. These early developmental alterations would then increase risk or enhance resiliency in relation to the development of PTSD in adults following an index trauma.
Temporal Model of Pathophysiology and Treatment Options for PTSD
Conceptualizing PTSD as a disorder of fear conditioning (
10,
127–
129) leads to the use of fear inhibition experiments to identify vulnerable and resilient individuals and can be used for testing treatments that bolster resilience.
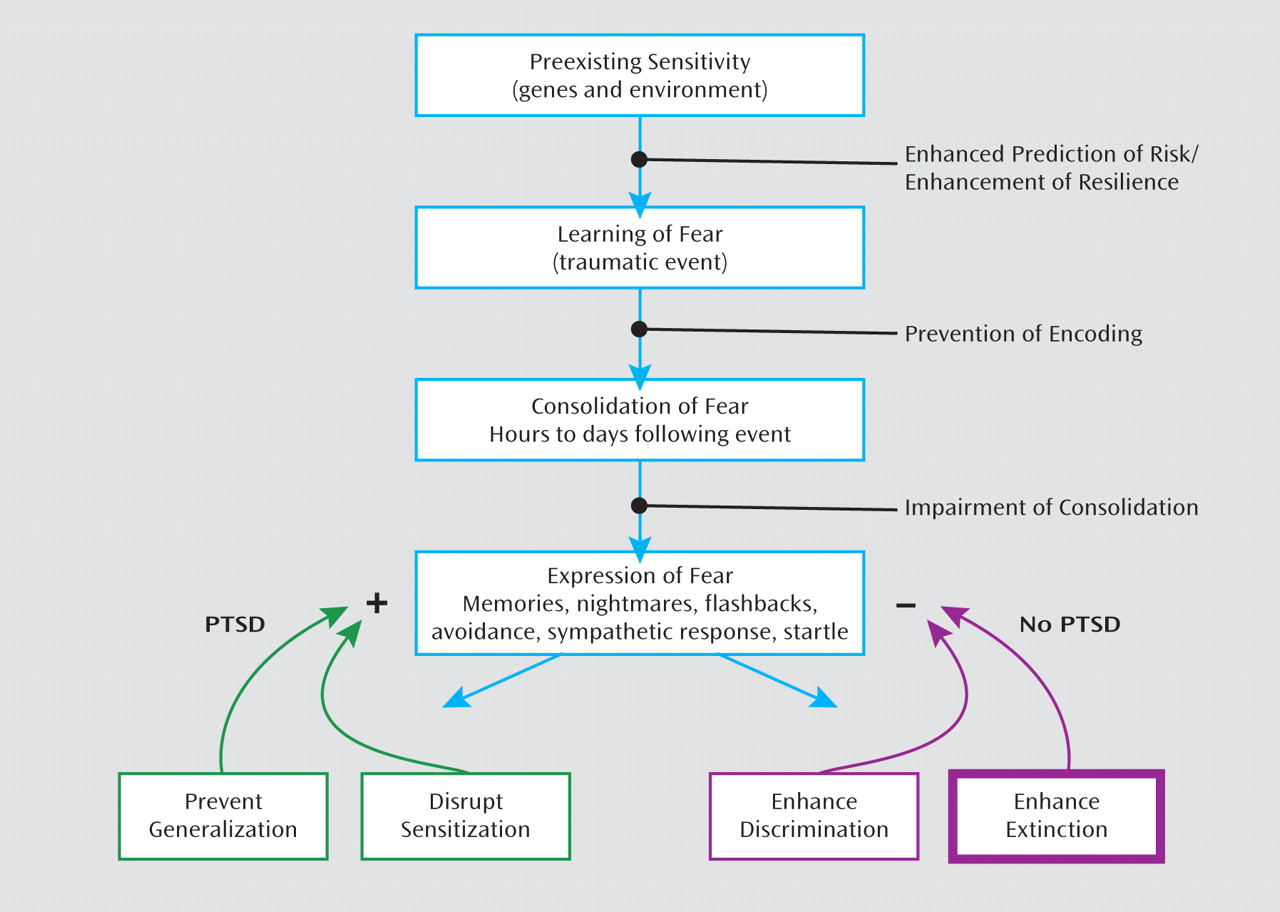
Figure 4 shows a putative timeline for the development of fear-related disorders, including PTSD. It starts with factors that increase vulnerability, such as genotype (for example, AA genotype of the
FKBP5 rs9296158 gene) and early life stress (such as childhood abuse). Exposure to trauma results in an association of all the external and internal stimuli present at the time of the event and the emotions of fear, horror, and helplessness that define the criteria of trauma. In an individual who will develop PTSD, these stimuli may later serve as reminders and cause symptoms of reexperiencing and hyperarousal. It is possible that intervention at this time, immediately following the event, can prevent the formation of a strong fear association and thus promote recovery. Several studies are under way in which victims of assault are treated in a hospital emergency department within hours after trauma exposure (
130).
Once the fear memory is formed, it can still be modified by methods that interfere with fear memory consolidation and its potential reconsolidation (
131). Consolidation and reconsolidation refer to the phenomenon in which a memory is repeatedly strengthened when trauma reminders become associated with hyperarousal symptoms; this results in a vicious circle by which fear memories lead to anxiety disorders. At this time point, it may be possible to intervene and modify the memory by associating it with safety rather than danger cues (
132,
133). If the fear memory is consolidated, trauma reminders will elicit expression of the fear response—that is, amygdala hyperactivity (
61). Exaggerated amygdala activity will be evident in the form of symptoms such as intrusive memories, nightmares, exaggerated startle responses (
9), and sympathetic nervous system activation, which increases heart rate, respiration rate, and sweating (
134). At this point in the course of the development of the disorder, individuals with healthy fear inhibition neurocircuitry might engage the prefrontal cortex to dampen amygdala activity. In fact, a new study (
134a) using an extinction paradigm that combined a single reactivation trial 10 minutes prior to extinction found that the fear memory was significantly reduced. More importantly, even a year after extinction, the fear memory was still reduced, indicating resistance to spontaneous recovery of fear. The authors argued that the fear memory, once reactivated, did not have an opportunity to be reconsolidated because of extinction. Treatments that employ mechanisms of fear inhibition, such as extinction, can potentially strengthen the inhibitory controls of the prefrontal cortex on the amygdala, thereby promoting recovery from PTSD.
Although most of this review has focused on mechanisms of fear inhibition and extinction, the failure to extinguish occurs when the excitatory memory of the trauma/CS+ outcompetes the inhibitory memory. Resistance to extinguishing in PTSD may therefore be due either to abnormally strong excitatory conditioning during acquisition or to impaired inhibitory conditioning during extinction. In sum, the confounding contribution of inhibitory and excitatory processes to abnormalities in conditioned inhibition and extinction in PTSD is critical. Figure 4 illustrates how differential memory processes can both inhibit and excite fear memory expression, with PTSD as a potential pathological outcome.
A type of treatment for PTSD that has its basis in extinction is exposure therapy. The term
exposure therapy refers to several behavioral and cognitive-behavioral treatment programs that involve confronting feared but safe thoughts, images, objects, situations, or activities in order to reduce pathological (unrealistic) fear, anxiety, and anxiety disorder symptoms. In the treatment of PTSD, exposure therapy usually involves prolonged imaginal exposure to the patient's memory of the trauma and in vivo exposure to various reminders of the trauma. This basic prolonged exposure protocol has been found to be highly effective in the treatment of women with PTSD following physical and sexual assault compared to waiting list or minimal attention control conditions (
135–
138). Similar exposure therapy programs have been successful with different trauma populations (
139–
141). Note that there are caveats to data on the prolonged exposure approaches to psychotherapy, however. Studies have often used limiting exclusion criteria and failed to address polysymptomatic presentations, which may render generalizability to a broader population of PTSD patients difficult to determine (
142).
Foa and Kozak (
143) suggested that two conditions are necessary for emotional processing to occur: activation of the fear memory and the incorporation of corrective information (e.g., that the feared consequence does not occur). These two conditions are met in exposure therapy when the patient confronts actual fear-related stimuli (in vivo exposure), intentionally creates an image of the feared situation, or intentionally retrieves a memory of the traumatic experience (imaginal exposure)
and experiences the associated fear reactions (indicated by self-reports of distress or physiological signs of arousal) but in the absence of the feared consequence (e.g., being assaulted). These processes are basically the same as those that occur in extinction, which can be studied directly in animals. Thus, repeated presentations of the CS (in vivo exposure—extinction training) typically elicits the fear response (activation), which then diminishes over the course of repeated trials within an extinction session as well as over the course of successive extinction sessions. Another similarity across exposure therapy and extinction training is a partial return of the conditioned response following exposure/extinction training at the beginning of the next session, referred to as the return of fear (
144) in the clinical literature and spontaneous recovery in the extinction literature. In addition, fear often returns in patients who undergo a subsequent major life stress (reinstatement) or even a change in context (renewal).
Pharmacology of Fear Inhibition
As the time course model for PTSD indicates (Figure 4), there may be several points at which therapeutic interventions can be done to prevent the development of PTSD. In addition to psychotherapy, several pharmacological approaches have been used at these time points. For instance, some data suggest that the administration of propranolol in the immediate aftermath of trauma may prevent fear consolidation (
145). Studies with animal models have shown that propranolol interferes with the formation of emotional memories (
146). Similarly, an early-intervention study of patients in an emergency department suggested that propranolol reduced the development of PTSD symptoms (
145). If replicated, these results may provide an exciting approach to preventing PTSD if early intervention is possible, although the time window for response may be limited. However, in most cases, too much time may have passed between the trauma and the treatment, particularly in cases where the trauma occurred in childhood or in combat, when immediate treatment was not available. In such cases, the fear memory is fully consolidated and administration of propranolol would no longer be effective. The most appropriate treatment in these cases would involve exposure therapy, which strengthens fear inhibition through extinction. Pharmacological agents that enhance safety learning would have an important application in this treatment approach.
A recent study showed that
D-cycloserine, a partial NMDA agonist, facilitated extinction when rats were tested drug free the next day (
147). These results have been fully replicated using freezing as the measure of conditioned fear, even when
D-cycloserine is administered up to 4 hours after extinction training (
148). This finding led to the first successful clinical test of combining
D-cycloserine with exposure-based psychotherapy (
149), a result that has since been replicated in several other studies (
150–
153). Remarkably, the rodent studies have shown that
D-cycloserine also seems to block later reinstatement (
154). In addition,
D-cycloserine leads to generalized extinction (
155), where extinction training to one cue in the presence of
D-cycloserine leads to a reduction of fear to another CS previously paired with the same US. This could be significant clinically because combining
D-cycloserine with exposure-based psychotherapy to specific cues associated with the original trauma might generalize to other cues associated with that traumatic event, even though these are not dealt with explicitly during therapy. A recent meta-analysis of more than 40 animal and human trials examining
D-cycloserine and extinction or exposure therapy concluded that
D-cycloserine "is a useful target for translational research on augmenting exposure-based treatment via compounds that impact neuroplasticity" (
156). Although no studies of the effects of
D-cycloserine in exposure therapy for PTSD have yet been completed, several are under way.
The pharmacological enhancement of extinction or, more specifically, the pharmacological enhancement of emotional learning that takes place during psychotherapy, is an increasingly interesting avenue of research. Notably, neuroimaging studies suggest that the prefrontal cortex regions engaged by top-down emotion regulation strategies may inhibit the amygdala (
24,
85). These connections may diminish fear through similar connections to the ventromedial prefrontal cortex that are thought to inhibit the amygdala during extinction. Also of note, one study suggested that orally administered
D-cycloserine may lead to inhibited amygdala activity during repeated presentation of faces (
157). A number of other avenues for enhancing extinction of fear are now being explored at the preclinical level. These include modulation of the cannabinoid system, which is known to influence local inhibitory circuits (
158–
160); modulation of GABA-ergic circuits directly (
161); modulation of BDNF-dependent neural plasticity (
74,
162); and enhancement of extinction through HPA axis modulation of cortisol (
163,
164).