“Selfish Spermatogonial Selection”: A Novel Mechanism for the Association Between Advanced Paternal Age and Neurodevelopmental Disorders
Abstract
Key Differences Between Male and Female Gamete Production
Advanced Paternal Age and Mental Disorders
Paternal Age Effect Mutations and Selfish Selection in the Testis
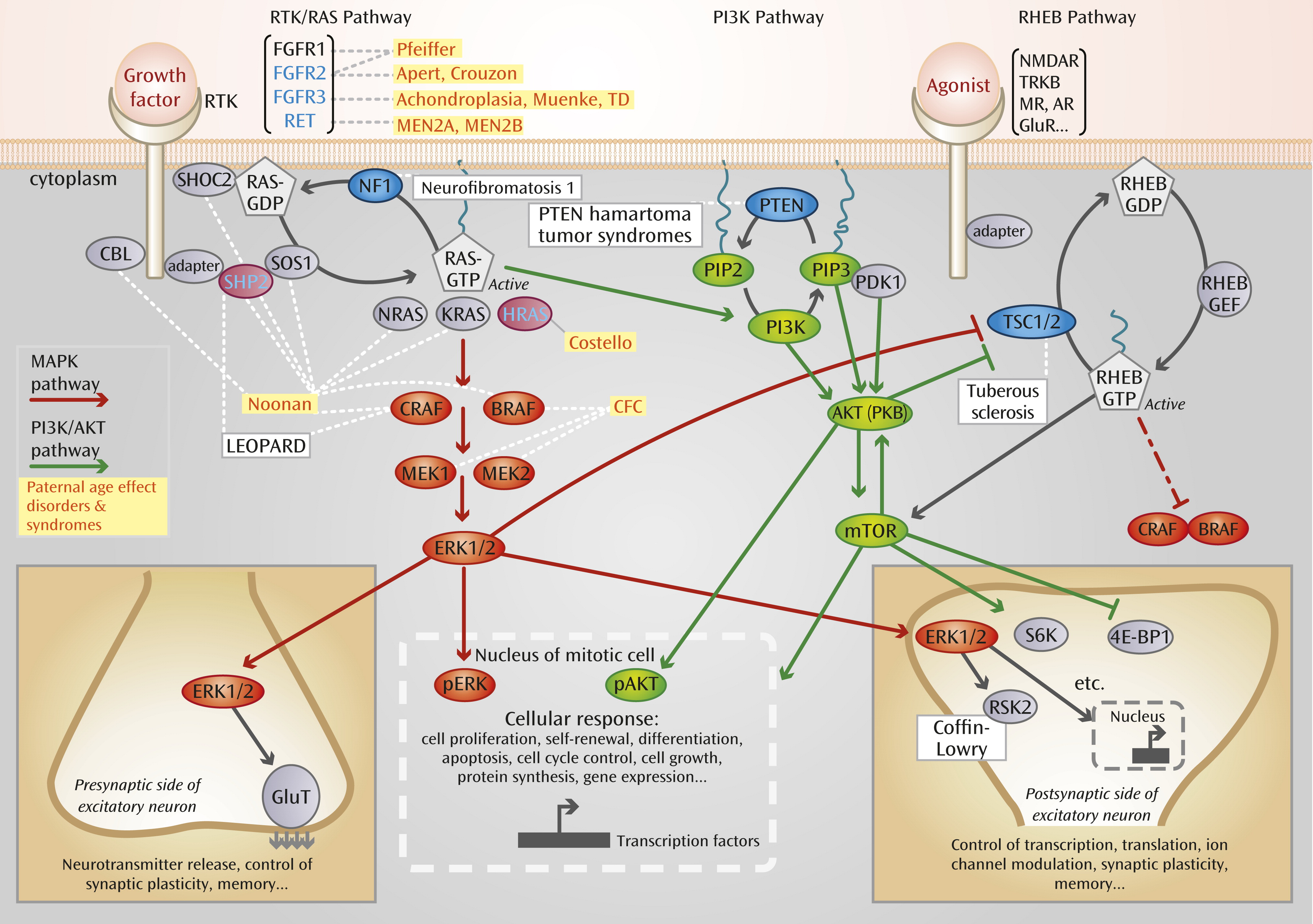
Genetic Architecture of Neurocognitive Disorders and Selfish Selection
Weak Selfish Mutations and Common Diseases
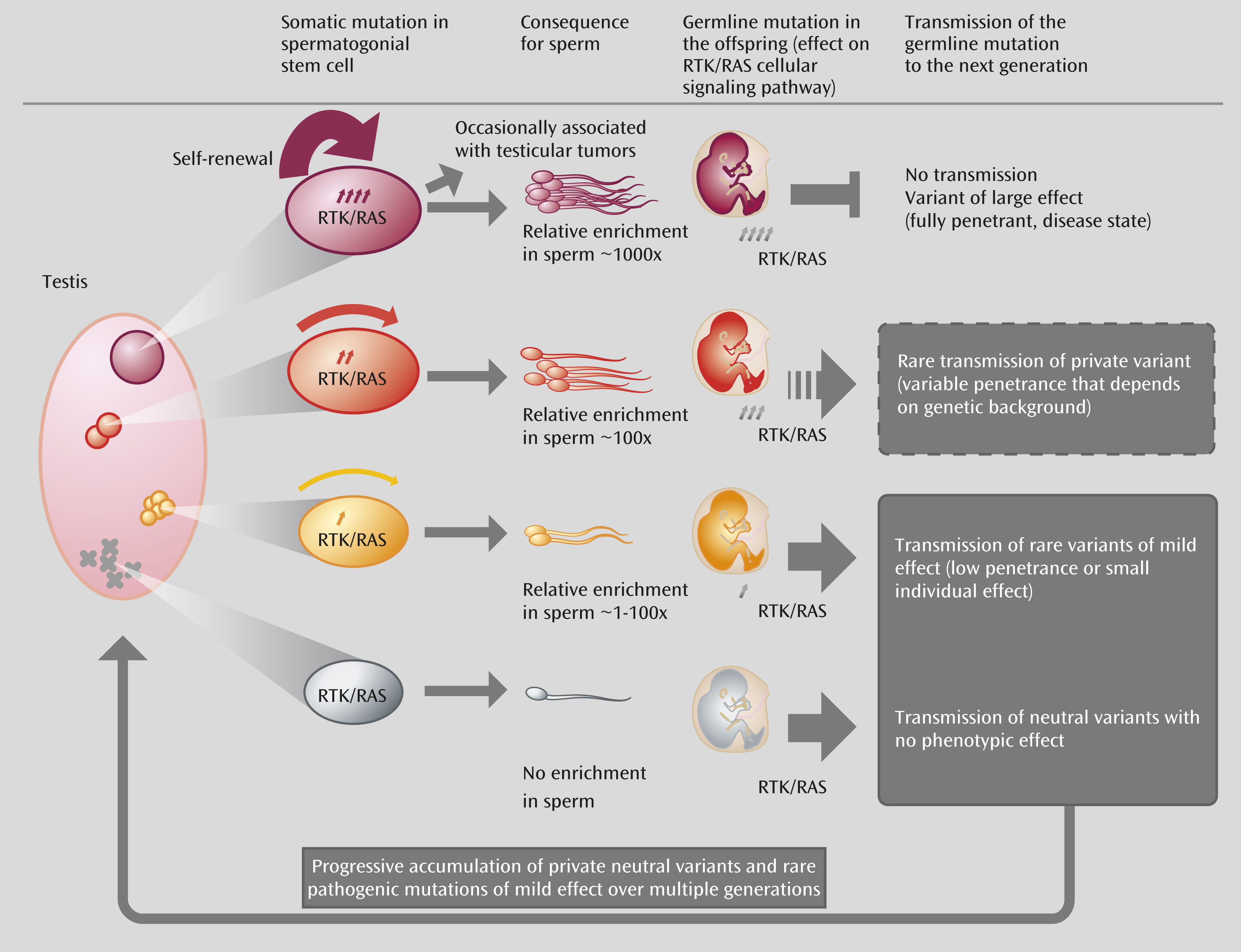
Does the Selfish Selection Hypothesis Have Explanatory Power?
Clues Linking Paternal Age, Cancer, and Neurodevelopmental Disorders
Clues Related to Minor Physical Anomalies, the Cell Cycle, and Synaptic Plasticity
An Integrative Model and Testable Hypotheses
Conclusions
Acknowledgments
References
Information & Authors
Information
Published In


History
Authors
Funding Information
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBLogin options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).