The human prion diseases are a group of rare neurodegenerative conditions. They share a common molecular pathological process, characterized by conversion of the normal cellular prion protein (PrP
c) into abnormal disease-associated forms (PrP
Sc), but their etiologies vary. They include sporadic Creutzfeldt-Jakob disease (sCJD), the inherited prion diseases (IPD), and the acquired prion diseases: variant CJD (vCJD), iatrogenic CJD (iCJD), and kuru (
1).
Clinically, these diseases are remarkably heterogeneous, both within and between the different disease types. As illustrated in the composite case vignette, they often cause complex neuropsychiatric syndromes that evolve rapidly through the course of the disease. Various factors have been shown to contribute to clinical heterogeneity in prion disease, including etiological disease type, prion strain, genotype at the polymorphic codon 129 of the prion protein gene (
PRNP), and demographic factors such as age (
2).
Behavioral and Psychiatric Features of Prion Disease
Behavioral and psychiatric symptoms have been recognized since the earliest clinical descriptions of prion disease (
3), but there has been relatively little research into their prevalence, phenomenology, natural history, and treatment. When vCJD first arose in the United Kingdom during the 1990s, early psychiatric symptoms were noted (
4,
5), and they are included in the World Health Organization diagnostic criteria for this disease (
6). A review of 106 pathologically confirmed cases of vCJD concluded that behavioral and psychiatric symptoms were present in 92% (
7). In 2005, Wall et al. (
8) reported a retrospective study of 126 sCJD cases evaluated at the Mayo Clinic over 25 years and found that psychiatric symptoms (excluding sleep disturbance) were present in 26% of cases at presentation, in 80% within the first 100 days after onset, and in 89% at any stage of their illness. Appleby et al. used retrospective case note review (
9) and meta-analysis of published cases (
10) to investigate patterns of symptoms in sCJD, and they suggest that there is a distinct “affective sCJD variant.”
Published experience of pharmacological treatments in prion disease has focused on putative disease-modifying agents. Interestingly, chlorpromazine and the tricyclic antidepressants are among a number of agents that have been found to inhibit prion replication in vitro (
11,
12), and their use in patients with prion disease has been described in a handful of case reports (
13–
16), but the reports address the failure of these agents to halt the progression of the disease rather than any usefulness in treating psychiatric symptoms. Beyond this, there has been no published evidence on which to base the symptomatic treatment of behavioral and psychiatric symptoms in patients with prion disease.
In contrast, behavioral and psychiatric symptoms in the common dementias have received increasing clinical and scientific attention in recent years. They are a major cause of morbidity in themselves, and they are associated with poorer outcomes for patients and more stress for caregivers (
17,
18). Large-scale research on their treatment has produced unexpected and important results in terms of both efficacy and safety (
19–
22) and has had a major impact on clinical practice. Several studies have used factor analysis to show that behavioral and psychiatric symptoms in Alzheimer’s disease tend to occur in a small number of clusters or “subsyndromes” (
23,
24). All studies have reported psychosis, mood disorder, and agitation/hyperactivity clusters.
In comparison with the more common dementias, there are a number of particular challenges to studying behavioral and psychiatric symptoms in prion diseases. Their rarity, their typically rapid progression, and a tendency for late diagnosis all make prospective study challenging. Expressive language dysfunction is a hallmark of prion disease, and this limits patients’ ability to describe the internal experience of psychiatric symptoms.
Prospective Data From U.K. Clinical Studies
The National Health Service’s National Prion Clinic offers specialist clinical assessment and follow-up for all cases of suspected prion disease in the United Kingdom. Over the past 10 years the clinic has also conducted two large clinical studies: the PRION-1 study and the ongoing National Prion Monitoring Cohort, which have achieved high levels of enrollment (>95% of eligible patients were enrolled in the Cohort). The PRION-1 study was an open-label patient preference trial of quinacrine for all types of prion disease, and the study showed no effect of quinacrine on survival or any of the rating scales used as secondary outcome measures (
25,
26); for our purposes here, the clinical data from the PRION-1 study are therefore regarded as natural history data. The National Prion Monitoring Cohort is a prospective natural history study. Full details of these studies have been published elsewhere (
25–
27).
In both the PRION-1 and National Prion Monitoring Cohort studies, details of symptoms at illness onset and at the time of each study assessment, along with the indications for any medication prescribed during the study period, were systematically recorded. The clinical case notes of selected patients identified from the research data, including all those for whom medication was prescribed for behavioral and psychiatric symptoms, were reviewed in detail. These studies provide valuable data on the psychiatric and behavioral features of the diseases, as well as systematically collected prospective clinical data on more than 350 patients (see
Table 1). Here we combine this experience with the previously published work to review the clinical characteristics, natural history, and in particular the treatment of these important symptoms of prion disease. Using other clinical and demographic data collected in these studies, we also assessed factors that predict the presence of particular behavioral and psychiatric symptoms, offering some insight into the pathophysiology underlying these clinical features.
As has been found previously in work on behavioral and psychiatric symptoms in dementia (
23,
24), clinical features occurring together in individual patients seemed to be divisible into three main groups: psychotic features (e.g., hallucinations, delusions); agitated features (e.g., agitation, aggression, behavioral disturbance); and mood disorder (e.g., low mood, emotional lability, apathy, withdrawal).
Psychotic Features
Prevalence and Pathophysiology
In our series, 36.9% of patients had psychotic features at any stage of their illness.
Table 2 summarizes the prevalence across different disease types. Psychotic features were particularly common in vCJD and sCJD. Using logistic regression analysis (with age at onset, gender, disease type, and
PRNP codon 129 genotype as covariates), presence of psychotic symptoms at any stage was independently predicted by specific disease types (sCJD > IPD) and codon 129 genotypes (MV > MM) with high levels of significance (p values <0.01) (see the
data supplement that accompanies the online edition of this article).
Phenomenology and Natural History
Psychotic features occurred with and without associated agitation or behavioral disturbance. By far the most common was visual hallucinosis, which occurred both in the context of other progressive visual symptoms (such as visual distortion and agnosia) and as an isolated phenomenon. Distressing visual distortions and illusions were also common. Several patients described unpleasant splitting and twisting of faces and other objects, “like a Picasso.”
In terms of hallucination content, there was a striking abundance of animals, and also human figures. In a few instances, patients described multimodal hallucinations (e.g., seeing, hearing, feeling, and smelling rats crawling over the body), but this was rare and was always associated with a particularly florid visual hallucinosis. In patients with more advanced disease, episodes characterized by a wide-eyed, staring, or fearful expression were frequently observed. Some of these patients had previously reported visual hallucinations at an earlier stage. A smaller number of patients were noted to have delusions, which tended to be relatively simple (e.g., theft or infidelity). Several patients reported vivid sensory experiences outside the limits of their real sensory field, such as “seeing” someone in another part of the house or a distant location—“extracampine” hallucinations—which were often associated with delusional thought content. Possible Schneiderian first-rank symptoms were noted in two patients with vCJD. A 54-year-old man with irritability, agitation, visual hallucinations, and myoclonus reported feeling “as if someone is taking over my mind,” and a 30-year-old man was noted to have “thought interference, delusional thinking, and possible hallucinations” before the onset of any frank neurological symptoms. Six of the 204 patients with sCJD had purely visual symptoms at onset (and could therefore be classified as having the Heidenhain variant of sCJD [
28]). They had symptoms such as loss of acuity, blurring, or distortions initially, but several developed hallucinations as the disease progressed. Another 29 sCJD patients had visual symptoms at onset (in combination with other symptoms), including hallucinations in 13 patients.
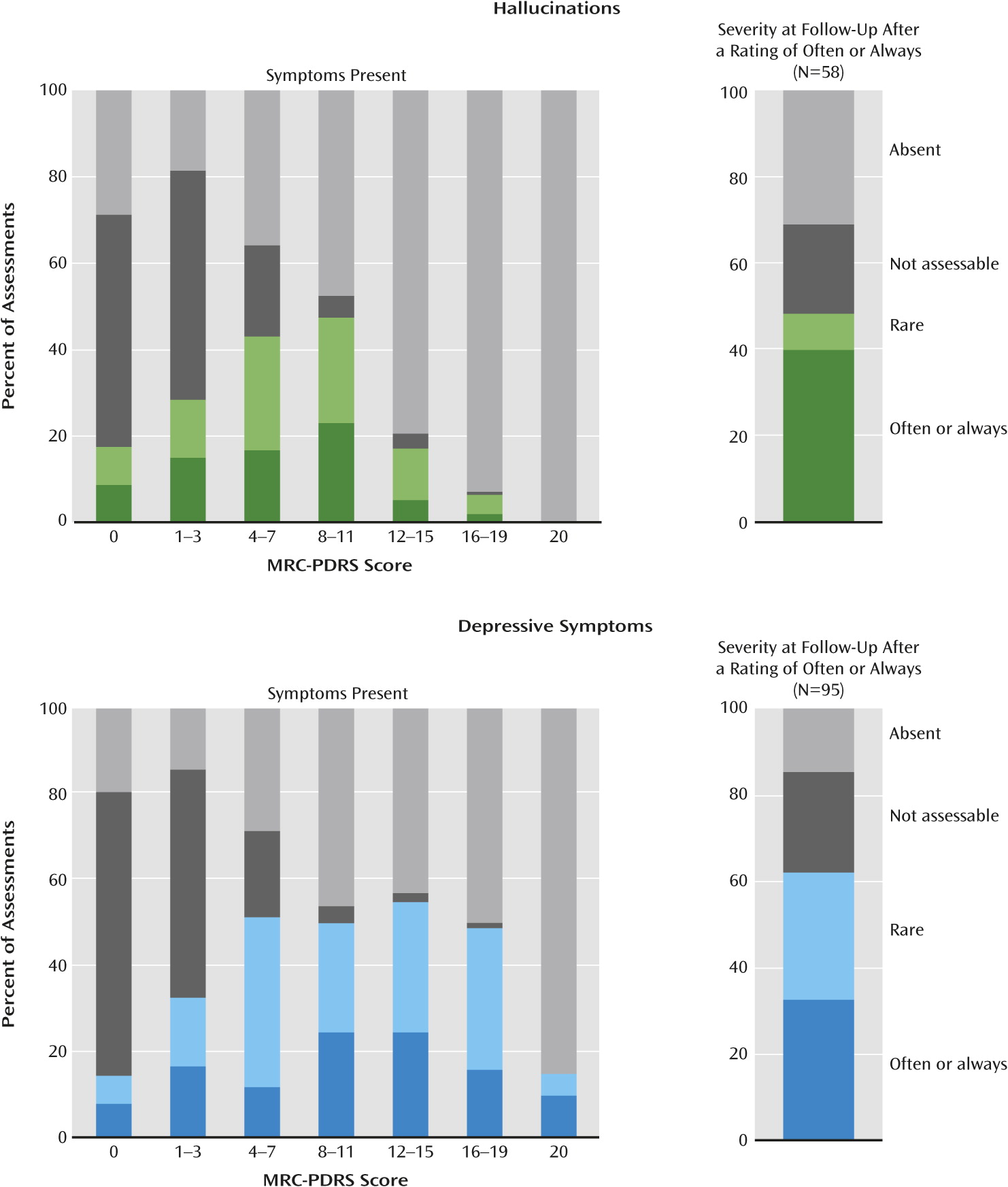
Figure 1 shows the proportion of assessments at which hallucinations were noted at different stages of disease progression, as assessed by the Medical Research Council Prion Disease Rating Scale (MRC-PDRS) (
27). Hallucinations were not observed in the most mildly impaired, and they became more prevalent as impairment increased. In keeping with this, logistic regression showed a highly significant effect of MRC-PDRS score on the odds of hallucinations being present (p<0.001), adjusted for disease type. To assess the natural history of these symptoms in individual patients,
Figure 1 also shows the severity of hallucinations recorded at the assessment following one at which they were rated “often” or “always.” The symptoms had improved in about 40% of cases, and in about 20% of cases, symptoms could no longer be assessed because of progression of the disease.
Pharmacological Treatment
Table 3 summarizes the observed pharmacological treatment of patients with behavioral and psychiatric symptoms in the PRION-1 and National Prion Monitoring Cohort studies.
A total of 31 prescriptions were written for antipsychotics for psychotic symptoms. The choice of specific agents was influenced by the risk of extrapyramidal side effects; quetiapine was the most frequently prescribed, and very few first-generation antipsychotics were used. Most treatment courses were short (median=61 days). In nine cases (29%), antipsychotics were noted to have apparent clinical benefit. Sedation was noted as an apparent adverse effect in six cases (19%); this often occurred in the context of severe neurological disability, and in no case was it recorded as a reason for discontinuation of the prescription.
There were 32 prescriptions of benzodiazepines for psychotic symptoms. Most were in the context of late-stage disease and were prompted by severe agitation or distress that seemed to be driven by psychotic symptoms. In a number of cases, myoclonus was noted as a joint indication. Most treatment courses were short (median=42 days). In seven cases (22%), benzodiazepines were noted to have apparent clinical benefit. Sedation was noted as an apparent adverse effect in four cases (13%) but was never recorded as a reason for discontinuation of the prescription.
Five prescriptions were written for acetylcholinesterase inhibitors for psychotic symptoms. The decision to use these agents was based on overlap between the neuropsychiatric symptomatology of some prion disease cases with that of Lewy body disease, in which these drugs are known to be effective in treating behavioral and psychiatric symptoms (
29,
30). Prescriptions were started in patients with moderately advanced disease, all of whom had hallucinations, and courses were relatively long (median=236 days). Three of the five (60%) prescriptions were noted to have apparent clinical benefit, and no adverse effects were recorded.
Agitated Features
Prevalence and Pathophysiology
Agitated (nonpsychotic) features were not systematically recorded at all study assessments, so the prevalences in
Table 2 are derived only from symptoms present at onset and symptoms requiring pharmacological treatment, and they are therefore likely to be significantly underestimated.
In logistic regression analysis, younger age at onset approached significance (in light of multiple testing involved) as an independent predictor of agitated features (odds ratio=0.966 per year, p=0.03) (see the online
data supplement).
Phenomenology and Natural History
Agitation and behavioral disturbance frequently occurred in the absence of psychotic features. Typical descriptions included “irritability,” “resistiveness,” and “hostility.” Other overactive behaviors, such as wandering and repetitive vocalizations, were also included. These symptoms were commonly noted to occur in response to external stimuli (most commonly administration of personal care). In some patients in the earlier stages of disease, nonpsychotic behavioral disturbance occurred in the context of disinhibition, impulsivity, and other signs of frontal lobe dysfunction. Patients with advanced disease often showed evidence of agitation without any behavioral evidence of hallucinations. Agitation was often reactive to stimulus and often coexisted with stimulus-sensitive myoclonus and an exaggerated startle response, which are common features in the late stages of all prion diseases. This was observed in some patients in whom agitated features had been noted earlier in the disease course, but also in many who did not have earlier agitation.
Pharmacological Treatment
A total of 23 prescriptions were written for antipsychotics to treat agitation (see
Table 3). These included very short treatment courses at the end stage of sCJD (minimum, 7 days) and much longer courses in slowly progressive IPD (maximum, 1,506 days). In 10 cases (43%), antipsychotics were noted to have apparent clinical benefit. Apparent adverse effects were noted in three cases (13%): extrapyramidal symptoms in two cases and sedation in one. None of these symptoms was severe, and they were not recorded as a reason for discontinuation.
There were 39 prescriptions for benzodiazepines for agitation, most initiated in late-stage disease. In a number of cases, myoclonus was noted as a joint indication. Most treatment courses were short (median=23 days). Six (15.4%) prescriptions were noted to have apparent clinical benefit. Sedation was noted as an apparent adverse effect in four cases (10.3%). This often occurred in the context of severe neurological disability, and it was not recorded as a reason for discontinuation of the prescription in any of the cases.
There were six prescriptions for antidepressants for agitation, in five patients with IPD treated at relatively early stages of disease, and treatment courses were long (median=750.5 days). Only one (17%) was noted to have apparent clinical benefit. Apparent adverse effects were noted in two cases (33%): weight gain and headache. The headache led to discontinuation of treatment.
Mood Disorder
Prevalence and Pathophysiology
Symptoms of mood disorder were noted in 41.6% of patients at any stage of disease.
Table 2 summarizes the variation in prevalence across disease types.
In logistic regression analysis, female gender approached significance as an independent predictor of mood disorder at any stage (odds ratio=1.68, p=0.049), but there were no other significant predictors (see the online
data supplement).
Phenomenology and Natural History
Some patients who were studied in the early stages of disease had symptoms of low mood combined with typical biological symptoms consistent with a “classical” depressive illness. Patients in the early stages of vCJD were often noted to have predominant symptoms of social withdrawal, irritability, anxiety, and low mood. Patients in the early to middle stages of disease were more commonly noted to have emotional lability. Patients in these stages were also often noted to have social withdrawal and apathy, albeit in the context of deteriorating expressive language and frontal executive function. Affective symptoms in patients in the late stages of disease could rarely be characterized.
Suicidal ideation was noted in a few cases, all in the early to middle stages of IPD. This varied from a patient with minimal cognitive impairment making inquiries regarding physician-assisted suicide to a patient with marked frontal deficits voicing suicidal intent impulsively and with incongruous affect. Only one suicide attempt was identified from the clinical case notes reviewed (a patient in the early stages of IPD), and there were no completed suicides.
As shown in Figure 1, depressive symptoms were observed at a similar rate across the range of functional impairment until the very severely impaired range, where they often could not be assessed. There was no significant effect of MRC-PDRS score on the odds of depressive symptoms being present. Figure 1 also shows the severity of depressive symptoms at the assessment following one at which they were rated “often” or “always.” The symptoms had improved in about 40% of cases, and in about 20% of cases, symptoms could no longer be assessed because of progression of the disease. Pharmacological Treatment
A total of 45 prescriptions were written for antidepressants for mood disorder (see
Table 3). Most of these were either in sCJD cases, where treatment courses tended to be shorter (median=111 days), or in IPD cases, where courses were longer (median=565 days). Sixteen prescriptions (36%) were noted to have apparent clinical benefit. Apparent adverse effects were noted in four cases (9%): agitation, sedation, weight gain, and a persistent unpleasant taste. None of these symptoms were severe, and in only one case (unpleasant taste) did the symptom lead to discontinuation.
Other Symptoms
Almost all symptoms noted could be classified into the three groups described above, but a few others did occur. Obsessive thoughts were reported in a handful of patients. Compulsive or repetitive behaviors were included in the agitated features group. One patient with IPD due to D178N mutation had a diagnosis of obsessive-compulsive disorder that predated onset of cognitive and neurological symptoms by many years. A recurrent sensation of “déjà vu” and a strong sensation of derealization were reported by two patients in the very early stages of sCJD.
Symptoms at the Onset of Illness
Behavioral and psychiatric symptoms arising very early in the disease course deserve particular attention. They are used as specific diagnostic markers for vCJD; patients may present to psychiatric services before they exhibit frank evidence of neurological disease and may be given an initial diagnosis of a primary psychiatric condition, as in our case vignette. In our series, 44.5% of patients had behavioral and psychiatric symptoms as part of their first symptoms of the disease, but only in 8.8% were they the only symptoms at this stage (see
Table 2). This highlights the importance of thorough assessment for neurological symptoms and signs in patients presenting to psychiatric services: the presence of neurological features should prompt further investigation for an organic cause, including prion disease and other neurodegenerative conditions.
As expected, early behavioral and psychiatric symptoms were particularly common in vCJD, with 92% of patients having such symptoms at illness onset (and 24% having only such symptoms at onset), and vCJD diagnosis was an independent predictor of the presence of behavioral and psychiatric symptoms at illness onset in logistic regression analysis (odds ratio=9.7, p=0.01). However, as sCJD is far more common than vCJD, most of the patients in the series with behavioral and psychiatric symptoms at onset had sCJD.
Analyzing sCJD patients separately using logistic regression (with age at onset, gender, PRNP codon 129 genotype, and distribution of pathological signal change on MR brain imaging as covariates), the presence of behavioral and psychiatric symptoms at onset was independently predicted by younger age at onset (odds ratio=0.935 per year, p=0.01), and no other strongly significant predictors were found. In a study using pooled data from a large number of published clinical cases, Appleby et al. (
10) also concluded that younger sCJD patients were more likely to present with particular symptoms, including affective symptoms and behavioral disturbance.
These findings raise the possibility that the presence of psychiatric symptoms at onset may (to some extent at least) be a feature of prion disease in younger people, as opposed to being primarily a feature of the vCJD etiological disease type. As the presence of early psychiatric features is currently used as a criterion for diagnosing vCJD (
7), this may have important implications: if older vCJD patients were less likely to exhibit this feature, it would be more difficult to distinguish them from the more numerous sCJD cases. This is of particular interest in the context of the evolving vCJD epidemic in the United Kingdom, where future patients presenting with longer incubation periods will tend to be older.
Safety of Antipsychotics
The use of antipsychotic medication in patients with dementia has been shown to be associated with a significant increase in mortality, partly explained by an excess of cerebrovascular events (
22,
31). It is important that we consider whether the same risks exist in patients with prion disease. Most antipsychotic treatment courses in our patients were relatively short, principally because of the rapid progression of disease. However, in an important minority of patients (mostly with IPD), progression is slower and treatment courses longer. Patients with prion disease are also younger than the dementia population in whom the risk has been demonstrated (the mean age of patients treated with antipsychotics in our series was 51.2 years). The baseline risk of cerebrovascular disease is therefore likely to be lower, and it seems likely that this would reduce the risk of this type of adverse event. However, our study was not powered to demonstrate a small change against the very high background mortality of a prion disease population, so a similar risk cannot be ruled out.
Recommendations
Behavioral and psychiatric features are common in all types of prion disease, and there is significant heterogeneity both within and between disease types. This should inform clinical care, as these symptoms represent a significant burden of morbidity for patients and caregivers but may easily go unreported and unrecognized unless clinicians are vigilant about them. The first step toward treating a symptom is to ask about it.
Most clinical features of prion disease result from the irreversible loss of brain functions and do not resolve once they have appeared. Our data show that behavioral and psychiatric symptoms, in contrast, fluctuate and improve in a substantial proportion of patients (in addition to many in whom manifest symptoms are short-lived because of rapid progression to akinetic mutism). This suggests that improving these symptoms with pharmacological or other interventions may be feasible, but also that improvement may occur without any specific intervention. It is important to bear this in mind when making decisions about treatment, interpreting the apparent effects of treatments, and designing clinical trials.
There has been no systematic study of nonpharmacological interventions for behavioral and psychiatric symptoms in prion disease, but from the case notes reviewed for our study and our clinical experience more generally, it is clear that such interventions play a vital role. The involvement of suitably trained and experienced caregivers and the care of patients in appropriate, calm, and containing settings seem to be particularly valuable. Providing full-time family caregivers with respite periods can greatly reduce levels of caregiver stress, and this should be offered if possible.
Although it is important to bear in mind that behavioral and psychiatric symptoms may respond to nonpharmacological measures and may improve spontaneously, we feel that there is a role for the judicious use of pharmacological treatments in some patients.
For more severe psychotic symptoms or agitation, short courses of antipsychotic medication appear to be effective in some cases. These cases should be reviewed regularly, and prescribers should be vigilant about adverse effects, such as sedation and extrapyramidal dysfunction, although in our experience adverse effects do not limit treatment in most cases. Longer treatment courses in slowly progressive forms of prion disease should be avoided if possible, in light of the safety concerns discussed above, and also because longer courses are not usually necessary. Benzodiazepines may be an appropriate option in the later stages of disease, where psychotic features often coexist with significant neurological disability and other distressing symptoms, such as myoclonus. Our limited experience of using acetylcholinesterase inhibitors for psychotic symptoms in prion disease seems promising, but the numbers are too small to allow any strong conclusions to be drawn.
The symptoms grouped as mood disorder above are likely to include several distinct clinical presentations—for example, patients in early disease stages who have a coexisting depressive illness and patients with more advanced dementia causing symptoms of emotional lability, apathy, or social withdrawal. The former may respond to standard treatments for depression, although cognitive impairment may limit patients’ ability to participate in standard talking therapies. Antidepressants may have a role in both situations, and our experience suggests that they are well tolerated in these patients.
It is important to note the limitations of the evidence presented here: the data related to treatment of symptoms is purely observational, the numbers are small, and the clinicians collecting the data were often the same people making treatment decisions. Nevertheless, in the absence of more definitive research, we hope our experience will provide a starting point for clinicians making treatment decisions with patients and caregivers and inspire further research into these challenging and distressing clinical features.
Acknowledgments
The authors thank all the individuals, their carers, and their families, who took part in the PRION-1 and Cohort studies, and U.K. neurologists and the National CJD Research and Surveillance Unit for referring patients. The authors gratefully acknowledge the contribution of current and past members of the National Prion Clinic, particularly Drs. Harpreet Hyare, Steve Wroe, Tom Webb, Suvankar Pal, Durre Siddique, Dilip Gajulapalli, Diego Kaski, and Christopher Carswell. They also thank Ray Young and Richard Newton for assistance with preparation of the figure and tables.