Bipolar I disorder is a serious, often disabling illness with an estimated prevalence of approximately 1%, an early age at onset (mean=18 years), and a chronic and recurrent course (
1,
2). Over the course of the illness, major depressive episodes predominate (
3,
4) and are associated with marked impairment in social and occupational functioning (
5), a heavy burden of both direct and indirect health care costs (
4–
6), and an increased risk of suicide (
7,
8). The World Health Organization (WHO) ranks bipolar disorder among the top 20 causes of disability worldwide (
9).
The chronicity and impact of depressive episodes associated with bipolar disorder, while greater than those of manic episodes, are not as well studied, and fewer treatment options are available (
10,
11). Currently, only two medications are approved in the United States, and in some other countries, for the treatment of acute bipolar depression: a combination of olanzapine-fluoxetine has demonstrated superiority to both olanzapine and placebo (
12), and quetiapine has demonstrated efficacy in both immediate and extended-release formulations (
13). Olanzapine has also demonstrated efficacy in bipolar I depression (
12,
14) and is approved for this use in Japan. The atypical agents aripiprazole and ziprasidone did not differentiate from placebo in randomized bipolar I depression trials (
15–
17).
Treatment with mood-stabilizing agents frequently provides insufficient antidepressant efficacy (
11,
18), and there is limited evidence supporting the use of standard tricyclic or serotonergic antidepressants for the treatment of bipolar depression (
10). Therefore, there is a substantial lack of effective and well-tolerated therapies for bipolar depression, underscoring the pressing need to identify proven new treatments for this condition.
Lurasidone is an atypical antipsychotic with high affinity for D
2, 5-HT
2A, and 5-HT
7 receptors (antagonist), moderate affinity for 5-HT
1A receptors (partial agonist), and no appreciable affinity for H
1 and M
1 receptors (
19,
20). Antagonist activity at 5-HT
7 receptors has been reported to be associated with antidepressant effects in animal behavioral models of depression (
21). Lurasidone has demonstrated significant improvement in both acute and chronic animal models of depression (
22). Furthermore, the antidepressant effect of lurasidone in these preclinical behavioral models was absent in 5-HT
7 knockout mice, indicating that this effect required the presence of functional 5-HT
7 receptors (
22). Therefore we hypothesized that the activity of lurasidone at 5-HT
7 and other 5-HT receptor subtypes make it a promising candidate as an antidepressant in patients with bipolar depression.
Lurasidone has been approved by the U.S. Food and Drug Adminstration for the treatment of bipolar I depression, both as monotherapy and as adjunctive therapy with lithium or valproate (
20). The primary objective of this placebo-controlled phase 3 study was to evaluate the efficacy of lurasidone monotherapy for the treatment of bipolar I depression.
Method
Subjects
This international study enrolled outpatients 18–75 years of age diagnosed with bipolar I disorder who were experiencing a major depressive episode (DSM-IV-TR criteria, ≥4 weeks and <12 months in duration), with or without rapid cycling, without psychotic features, and with a history of at least one lifetime bipolar manic or mixed manic episode. Diagnosis was confirmed by the Mini-International Neuropsychiatric Interview (
23) and the Bipolarity Index (
24). A Montgomery-Åsberg Depression Rating Scale (MADRS [
25]) score ≥20 and a Young Mania Rating Scale (YMRS) score ≤12 were required at both screening and baseline.
Patients were excluded if they demonstrated a decrease of ≥25% in MADRS total score between screening and baseline; scored ≥4 on MADRS item 10 (suicidal thoughts) at screening or baseline; had a history of nonresponse to an adequate (6-week) trial of three or more antidepressants (with or without mood stabilizers) during the current depressive episode, or demonstrated imminent risk of suicide or injury to self, others, or property.
The study was approved by an institutional review board at each investigational site and was conducted in accordance with the International Conference on Harmonisation Good Clinical Practices guidelines and with the ethical principles of the Declaration of Helsinki. All patients who entered the study reviewed and signed an informed consent document explaining study procedures and potential risks before study entry. An independent data and safety monitoring board reviewed and monitored subject data throughout the study.
Study Design
This randomized, double-blind, placebo-controlled, fixed-flexible dose, parallel-group monotherapy study of lurasidone compared with placebo was conducted between April 2009 and February 2012. A total of 505 patients underwent random assignment at sites in the United States (205 patients from 24 sites), India (76 patients from nine sites), Ukraine (52 patients from five sites), Czech Republic (55 patients from four sites), South Africa (57 patients from four sites), Russia (31 patients from four sites), Romania (16 patients from four sites), and France (13 patients from one site). Following a washout period of at least 3 days, patients were randomly assigned in a 1:1:1 ratio via an interactive voice response system to receive 6 weeks of treatment with lurasidone, at flexible daily doses of either 20–60 mg or 80–120 mg, or 6 weeks of placebo. Study medication was provided in blister packs as either lurasidone 20 mg or 40 mg, or identically matched placebo tablets, and was taken once daily in the evening, with a meal or within 30 minutes after eating.
Patients assigned to receive lurasidone 20–60 mg/day were treated with 20 mg/day for days 1–7. Patients assigned to the lurasidone 80–120 mg/day arm received a fixed titration of lurasidone as follows: 20 mg/day for days 1–2, 40 mg/day for days 3–4, 60 mg/day for days 5–6 and 80 mg/day on day 7. In both treatment arms, lurasidone dosing adjustments within the assigned dosing range were permitted after 7 days to optimize efficacy and tolerability. Lurasidone (or placebo) was taken once daily in the evening, with a meal or within 30 minutes after eating.
Concomitant Medications
Treatment with anticholinergic agents, propranolol, or amantadine, was permitted as needed (but not prophylactically) for movement disorders. Lorazepam, temazepam, or zolpidem (or their equivalent) were permitted during screening and for weeks 1 to 3, as needed for anxiety or insomnia, but not prophylactically, and not within 8 hours of any psychiatric assessment.
Efficacy Assessments
Efficacy assessments were obtained at baseline and weekly intervals. The primary efficacy endpoint was the mean change from baseline to week 6 in MADRS total score. At each study visit, a qualified site-based rater conducted the MADRS assessment; a second MADRS assessment was administered and scored by computer as part of a quality control process (Concordant Rater Systems, Boston, MA). The key secondary efficacy endpoint was the mean change from baseline to week 6 in depression severity score on the Clinical Global Impressions scale for use in bipolar illness (CGI-BP [
26]).
Additional secondary efficacy assessments included the 16-item Quick Inventory of Depressive Symptomatology (
27), the Hamilton Anxiety Rating Scale (HAM-A [
28]), the Sheehan Disability Scale (
29), and the Quality of Life Enjoyment and Satisfaction Questionnaire–Short Form (
30).
Safety and Tolerability Evaluations
Safety and tolerability were assessed by the incidence and severity of adverse events during the study. Movement disorders were assessed by the Simpson-Angus Scale, the Abnormal Involuntary Movement Scale, and the Barnes Akathisia Rating Scale. Additional safety evaluations included vital signs, laboratory tests, 12-lead ECG, and physical examination. Treatment-emergent mania was defined a priori as a Young Mania Rating Scale (YMRS [
31]) score ≥16 on any two consecutive visits, or at the final assessment, or an adverse event of mania or hypomania. Suicidal ideation and behavior were assessed using the Columbia Suicide Severity Rating Scale (
32).
Statistical Analysis
The intent-to-treat population consisted of randomly assigned subjects who received at least one dose of study medication and had at least one postbaseline efficacy assessment. The primary (MADRS) and key secondary (CGI-BP depression severity) efficacy endpoints were assessed using a mixed model for repeated-measures analysis including treatment, visit, pooled center, baseline score, and a treatment-by-visit interaction term, using an unstructured covariance matrix for within-subject correlation. The Hommel-based tree gatekeeping procedure was applied to control the family-wise type I error rate at 5% by taking into account the primary and key secondary efficacy variables and multiple dose comparisons of lurasidone versus placebo. There was no multiplicity adjustment for the secondary efficacy analyses or safety analyses.
Change from baseline in secondary efficacy measures was evaluated using an analysis of covariance (ANCOVA) model with fixed effects for treatment, pooled center, and baseline score as a covariate. Core depressive symptoms were evaluated using the MADRS-6 subscale (
33). The proportions of responders (≥50% reduction from baseline in MADRS total) and remitters (MADRS total ≤12) were compared between the lurasidone and placebo treatment groups using logistic regression. Cohen’s d effect sizes were calculated for the primary outcome as the difference in the change score divided by the pooled standard deviation. The number needed to treat to attain an additional responder was derived for the lurasidone group as follows: number needed to treat=1/(lurasidone response rate−placebo response rate).
The safety population included all subjects who were randomly assigned and received at least one dose of study medication. Descriptive statistics were used for safety variables including adverse events, vital signs, and laboratory results. In addition, a nonparametric rank ANCOVA was used to analyze weight and labaoratory parameters. Change from baseline to last observation carried forward endpoint in the Simpson-Angus Scale, the Abnormal Involuntary Movement Scale, and the Barnes Akathisia Rating Scale scores were analyzed by using an analysis of covariance (ANCOVA) model with fixed effects for treatment, pooled center, and baseline score as a covariate.
Sample size was determined by using an SAS simulation macro (
34) and was powered at 82.4% to reject both null hypotheses of no difference in MADRS between placebo and lurasidone dosage groups (3.5-point difference; pooled standard deviation of 9), and was powered at 96% to reject at least one null hypothesis of no difference from placebo for the lurasidone dosage groups. The estimated sample size of N=168 per treatment arm (total N=504) included an additional eight patients (5%) per arm based on expected early attrition (patients randomly assigned but without any postbaseline efficacy measures).
Results
Patients and Disposition
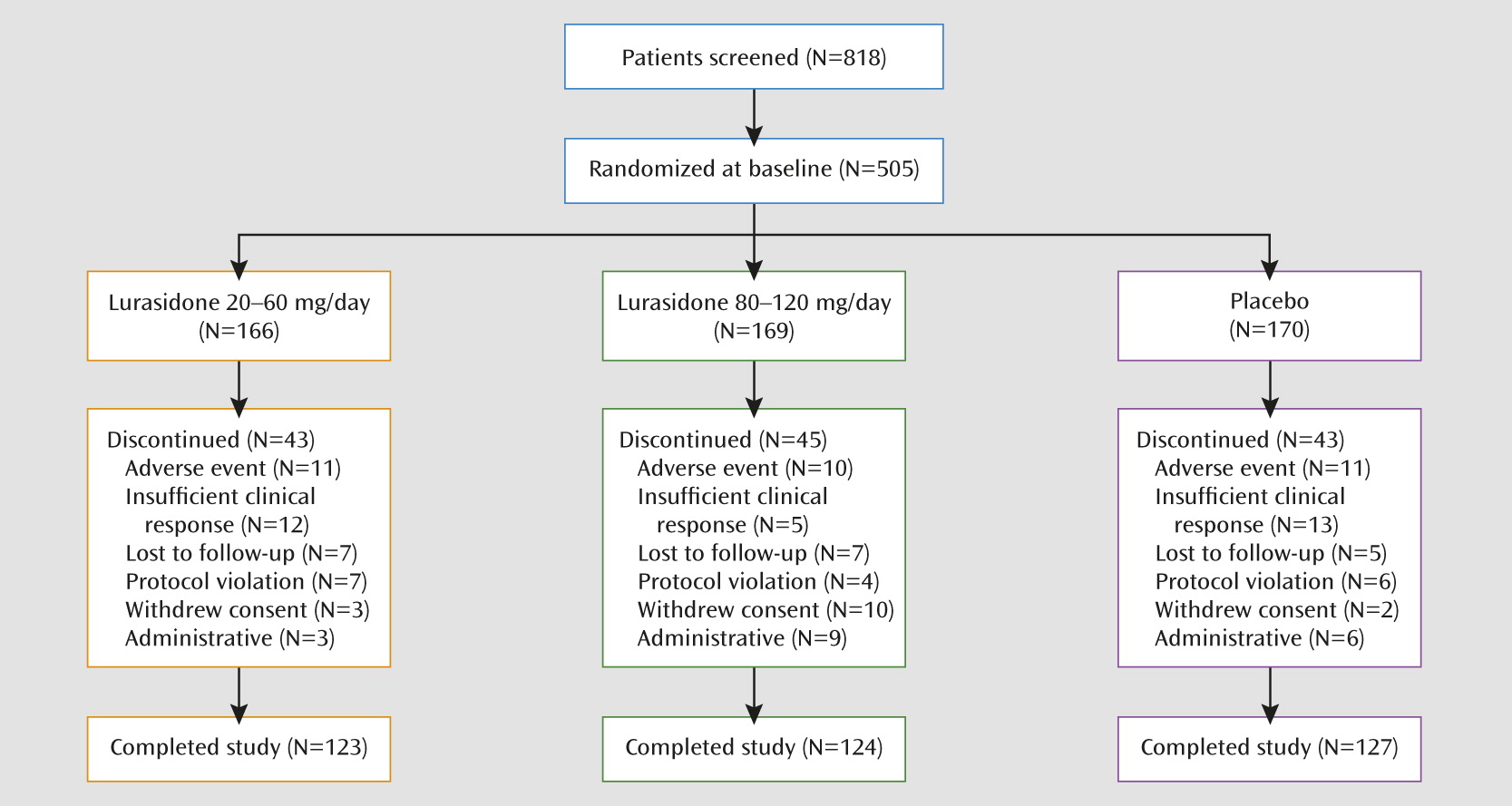
A total of 818 patients were screened, of whom 505 (61.7%) were randomly assigned to 6 weeks of double-blind treatment (
Figure 1); 499 received at least one dose of study medication (safety population), and 485 had at least one postbaseline efficacy assessment (intent-to-treat population). Baseline demographic and clinical characteristics were similar for the two treatment groups (
Table 1). Study completion rates were similar for the lurasidone 20–60 mg (74%), lurasidone 80–120 mg (73%), and placebo (75%) groups (
Figure 1).
The mean daily dose of lurasidone during the study was 31.8 mg in the 20–60 mg group and 82.0 mg in the 80–120 mg group. For the 20–60 mg group (after 20 mg/day for 7 days), 66.7% of patients increased to a dosage of 40 mg/day, and 34.0% increased to a dosage of 60 mg/day at some point during the remaining 5 weeks of study treatment. For the 80–120 mg group (after fixed dose titration from 20 mg/day to 80 mg/day at day 7), 62.0% of patients increased to a dosage of 100 mg/day, and 26.6% increased to a dosage of 120 mg/day at some point during the remaining 5 weeks of study treatment.
As-needed treatment with lorazepam was reported by 12% of patients in the lurasidone 20–60 mg group, 13% of patients in the lurasidone 80–120 mg group, and 19% of patients in the placebo group. As-needed treatment with zolpidem was reported by 13% of patients in the lurasidone 20–60 mg group, 8% of patients in the lurasidone 80–120 mg group, and 8% of patients in the placebo group.
Efficacy
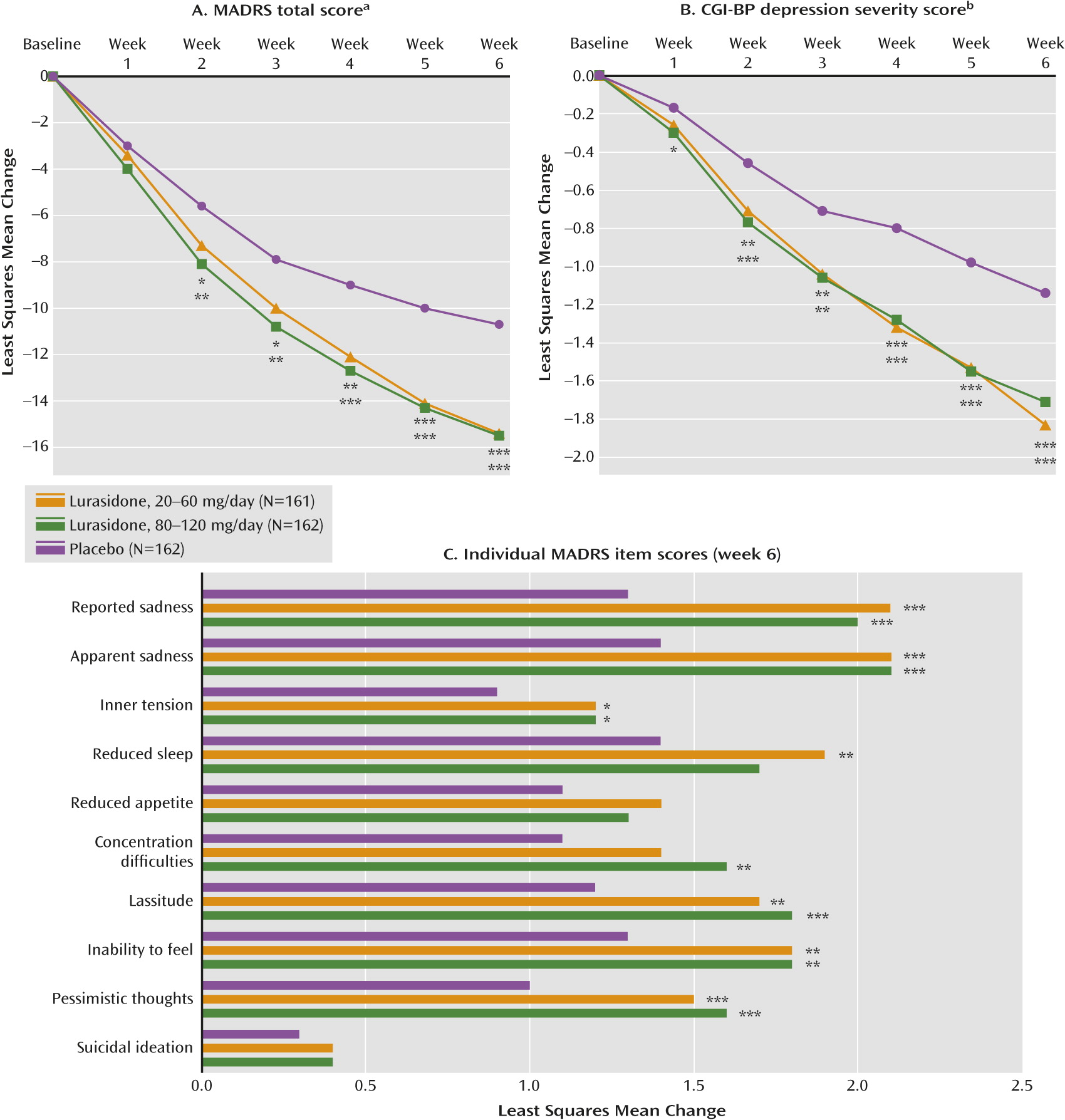
The least squares mean change from baseline to week 6 in MADRS total score (the primary endpoint) was significantly greater than seen with placebo (−10.7) for the lurasidone 20–60 mg group (−15.4; p<0.001 [effect size=0.51]) and the lurasidone 80–120 mg group (−15.4; p<0.001 [effect size=0.51]) (
Figure 2A). The least squares mean change from baseline to week 6 in CGI-BP depression severity score (key secondary endpoint) was significantly greater than seen with placebo (−1.1) for the lurasidone 20–60 mg group (−1.8; p<0.001 [effect size=0.61]) and the lurasidone 80–120 mg group (−1.7; p<0.001 [effect size=0.50]) (
Figure 2B). For both dosages of lurasidone, superiority compared with placebo on the MADRS was observed starting at week 2 and was maintained at all subsequent study visits. Superiority compared with placebo on the CGI-BP depression severity score was observed starting at week 1 for the lurasidone 80–120 mg group and starting at week 2 for the lurasidone 20–60 mg group and was maintained at all subsequent study visits.
There was a statistically significant reduction from baseline to week 6 in core depressive symptoms (MADRS-6 subscale score) for the lurasidone 20–60 mg group (−10.4; p<0.001) and the lurasidone 80–120 mg group (−10.4; p<0.001) relative to the placebo group (−6.9). Lurasidone was associated with significantly greater improvement than placebo on seven of the 10 MADRS items in both the 20–60 mg and 80–120 mg groups (
Figure 2C).
A significantly greater proportion of subjects met a priori response criteria after 6 weeks of treatment with lurasidone 20–60 mg (53%; p<0.001 [number needed to treat=5]) and lurasidone 80–120 mg (51%; p<0.001 [number needed to treat=5]) compared with placebo (30%). Median time to response was shorter in the lurasidone 20–60 mg group (34 days) and the 80–120 mg group (30 days) compared with the placebo group (42 days; log-rank p<0.01 for both comparisons).
The proportion of subjects achieving remission at endpoint was significantly greater in the lurasidone 20–60 mg group (42%; p=0.001 [number needed to treat=6]) and the lurasidone 80–120 mg group (40%; p=0.004 [number needed to treat=7]) compared with the placebo group (25%).
No significant treatment interactions by gender, age, race, or ethnicity were observed for either the MADRS total score or the CGI-BP depression severity score, based on ANCOVA analyses.
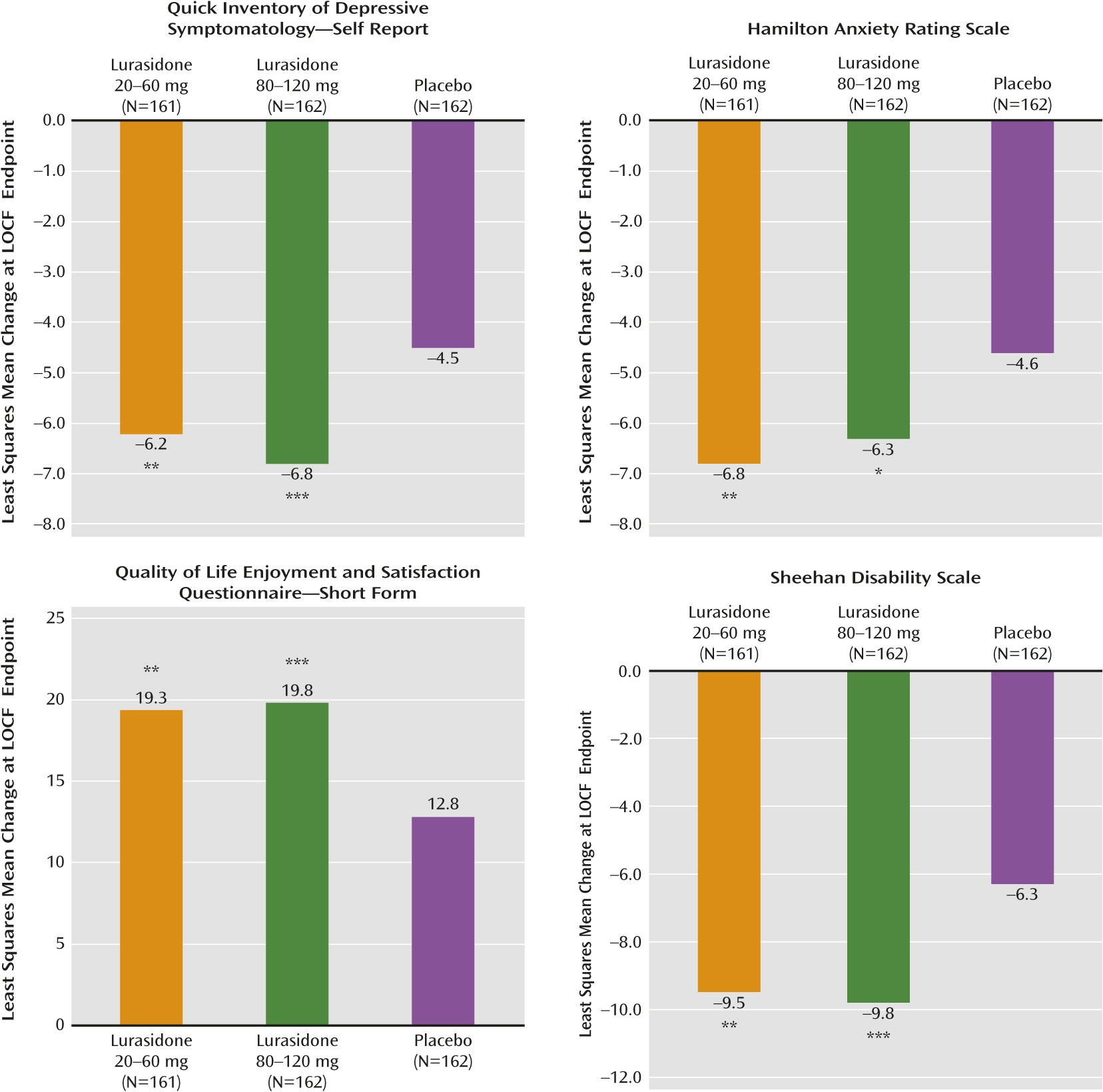
Treatment with both dosages of lurasidone was associated with significant improvement compared with placebo in anxiety symptoms, as measured by the clinician-rated Hamilton anxiety scale, the patient-rated Quick Inventory of Depressive Symptomatology, the Quality of Life, Enjoyment, and Satisfaction Questionnaire, and the Sheehan Disability Scale (
Figure 3).
Safety
Overall, adverse events reported with an incidence ≥5% in at least one of the groups are listed in
Table 2. The majority of adverse events were considered mild or moderate, with <10% rated as “severe” in the lurasidone 20–60 mg (9.8%) and 80–120 mg (8.4%) groups or the placebo group (6.0%). The incidence of serious treatment-related adverse events was low and similar in the lurasidone 20–60 mg group (N=2), the 80–120 mg group (N=1), and the placebo group (none). No deaths occurred during the study. The proportion of patients who discontinued due to adverse events was similar in the lurasidone 20–60 mg group (6.6%), the 80–120 mg group (5.9%), and the placebo group (6.5%;
Figure 1).
Treatment-emergent mania occurred in 3.7% of patients in the lurasidone 20–60 mg group, 1.9% in the lurasidone 80–120 mg group, and 1.9% in the placebo group. Mean YMRS scores showed a similar endpoint reduction in the lurasidone 20–60 mg group (−1.1), the 80–120 mg group (−0.7), and in the placebo group (−0.9).
The proportion of patients with at least one instance of treatment-emergent suicidal ideation, per the Columbia Suicide Severity Rating Scale, was 13.7% in the lurasidone 20–60 mg group, 14.8% in the lurasidone 80–120 mg group, and 13.6% in the placebo group. There was no occurrence of suicidal behavior or completed suicide during the study.
The incidence of extrapyramidal symptom-related adverse events was less than 10% in both lurasidone groups, with a modest dose-related increase in incidence (
Table 2). The proportion of patients who received treatment with anticholinergic medication for acute extrapyramidal symptoms was 3.7% in the lurasidone 20–60 mg group, 4.9% in the lurasidone 80–120 mg group, and 1.9% in the placebo group. Least squares mean changes from baseline to endpoint (lurasidone 20–60 mg and 80–120 mg versus placebo) were small for the Barnes Akathisia Scale (0.0 and 0.2 versus −0.1), and for the Simpson Angus Scale (0.02 and 0.02 versus 0.00). There were no significant changes from baseline to endpoint in the Abnormal Involuntary Movement Scale total score in any treatment group with no statistically significant differences between the lurasidone treatment groups and the placebo group.
There were no notable differences in laboratory measures, vital signs, or ECG assessments between treatment groups. Mean endpoint change in weight was not different for the lurasidone 20–60 mg group (+0.6 kg), the lurasidone 80–120 mg group (0.0 kg), and the placebo group (0.0 kg). The proportion of subjects with ≥7% increase in weight at endpoint was 4.2% in the lurasidone 20–60 mg group, 0.7% in the 80–120 mg group, and 0.7% in the placebo group. Mean change in waist circumference was also similar in the combined lurasidone group compared with the placebo group (0.1 cm versus −0.3 cm).
There were no clinically meaningful differences in the effect of treatment on metabolic parameters between the lurasidone groups and the placebo group (
Table 3). Median endpoint change in prolactin for lurasidone 20–60 mg and 80–120 mg, respectively, versus placebo was higher in female than in male subjects. The mean endpoint change in QTcF interval from baseline to endpoint was 0.5 ms in the lurasidone 20–60 mg group, −1.0 ms in the lurasidone 80–120 mg group, and −1.7 ms in the placebo group. No patient in either lurasidone group had a postbaseline change in QTcF>60 ms, and no patient in either lurasidone group had a QTcF>500 ms.
Discussion
In this multicenter, placebo-controlled, short-term study, monotherapy with lurasidone, in the fixed-flexible dosage ranges of 20–60 mg/day and 80–120 mg/day, significantly improved depressive symptoms in patients with bipolar depression. For both daily dose ranges, lurasidone was associated with significantly greater improvement in the MADRS starting at week 2 and at all subsequent visits through the week 6 study endpoint. These findings were supported by significantly greater improvement in the key secondary outcome, CGI-BP depression severity, which assessed the global severity of depressive symptoms, and by significant improvement in the core symptoms of depression as assessed by the MADRS-6 subscale. In addition, treatment with both dosage ranges of lurasidone was associated with significant improvement in anxiety symptoms, functional impairment, and quality of life. There were no significant treatment interactions by gender, age, race, or ethnicity.
A fixed-flexible dose design was selected to allow some of the advantages of flexibility in dosing while still permitting assessment of dose-response relationships (
35). The present study found comparable efficacy for the 20–60 mg/day and 80–120 mg/day dosage ranges of lurasidone, suggesting that treatment should be started at 20 mg/day and increased as needed over the 20–120 mg/day range (
20).
The magnitude of improvement in MADRS scores at week 6 in the lurasidone treatment groups was comparable to results from previous placebo-controlled monotherapy trials with atypical antipsychotics, as summarized in a recent meta-analysis (
36). The lurasidone versus placebo difference in MADRS scores was 4.6 for both dosage ranges in the current study, while the weighted mean drug versus placebo difference in MADRS scores in the meta-analysis ranged from 1.1 for aripiprazole and 3.1 for olanzapine to 4.6 for quetiapine 600 mg (immediate-release only) and 4.8 for quetiapine 300 mg (extended- and immediate-release [
37]). Since this meta-analysis was completed, an additional large placebo-controlled study of olanzapine has been reported, with a mean difference (versus placebo) on the MADRS of 2.2 (
14).
Lurasidone has been extensively studied for the short- and long-term treatment of schizophrenia (
20), and the present study revealed no new safety concerns or risks. Consistent with previous studies, treatment with lurasidone was associated with a small dose-related effect on prolactin, and minimal changes in weight, lipids, and measures of glycemic control. Lurasidone has been reported to have appreciably fewer weight and metabolic effects than other atypical antipsychotic agents (
37). The diagnosis of bipolar disorder is associated with high rates of metabolic syndrome (
38) and a significant increase in the risk of cardiovascular disease (
39). The metabolic profile of lurasidone reported here is consistent with prior schizophrenia studies and suggests that it may be associated with low cardiometabolic risk in this vulnerable clinical population.
No difference was observed between the lurasidone groups and placebo in the incidence of either treatment-emergent mania or suicidal ideation. No suicidal behavior occurred. Mania symptom severity was low at baseline as measured by the YMRS and showed small reductions over the course of the study in all treatment groups.
Treatment with both dosage ranges of lurasidone was well tolerated, with discontinuation rates due to adverse events that were low and similar to placebo. There was a modest, dose-related increase in the frequency of nausea, sedation, vomiting, and extrapyramidal symptoms for the 80–120 mg/day range versus the 20–60 mg/day range. Adverse events were typically mild to moderate in intensity, with <10% of events in the lurasidone groups rated as “severe,” only slightly higher than the incidence reported with placebo.
Several study limitations should be noted. Since only patients with bipolar I depression were enrolled, the extent to which these findings can be generalized to patients with bipolar II depression is not clear. Study entry criteria that excluded patients with serious psychiatric or medical comorbidity, and active suicidal ideation or behavior, also may have reduced the generalizability of the results. In this study, the suicidal thoughts item of the MADRS did not separate from placebo, perhaps in part because of low baseline severity. Further investigation is needed to evaluate the maintenance efficacy and longer-term safety of lurasidone in patients with bipolar disorder.
In conclusion, the results of this study found lurasidone, in the dosage range of 20–120 mg, to be an efficacious treatment for bipolar depression, with improvements observed in depressive and anxiety symptoms, functional impairment, and quality of life. Treatment with lurasidone was well tolerated, with minimal effect on weight, lipid parameters, and measures of glycemic control. This clinical profile suggests that lurasidone may be a valuable addition to the therapeutic armamentarium for the treatment of patients with bipolar depression.
Acknowledgments
The authors thank the participants of this study, as well as the members of the Lurasidone Bipolar Disorder Study Group (Czech Republic: Drs. J. Drahozal, E. Herman, M. Klabusayova, and Z. Solle; France: Dr. M. Zins Ritter; India: Drs. M. Chudgar, H. Gandhi, M. Gowda, V. Indla, U. Nagapurkar, J. Trivedi, P. Thatikonda, M. Vaishnav, and R. Yadav; Romania: Drs. M.G. Badescu, D. Cozman, G. Oros, and D. Vasile; Russia: Drs. M. Ivanov, V. Kozlovsky, V. Tochilov, and V. Vid; South Africa: Drs. K. Botha, G. Jordaan, L. Nel, J. Schronen; Ukraine: Drs. V. Abramov, V. Bitenskyy, S. Rymsha, A. Skrypnikov, and V. Verbenko; United States: Drs. S. Aaronson, M. Alam, S. Atkinson, P. Bhatia, R. Brenner, J. Calabrese, A. Cutler, G. Dempsey, D. Garcia, E. Gfeller, H. Hassman, R. Knapp, R. Manning, J. Miller, N. Rosenthal, R. Hidaldo, T. Tran-Johnson, N. Vatakis, D. Walling, R. Weisler, K. Yadalam, M. Allen, J. Kunovac, and S. Potkin).