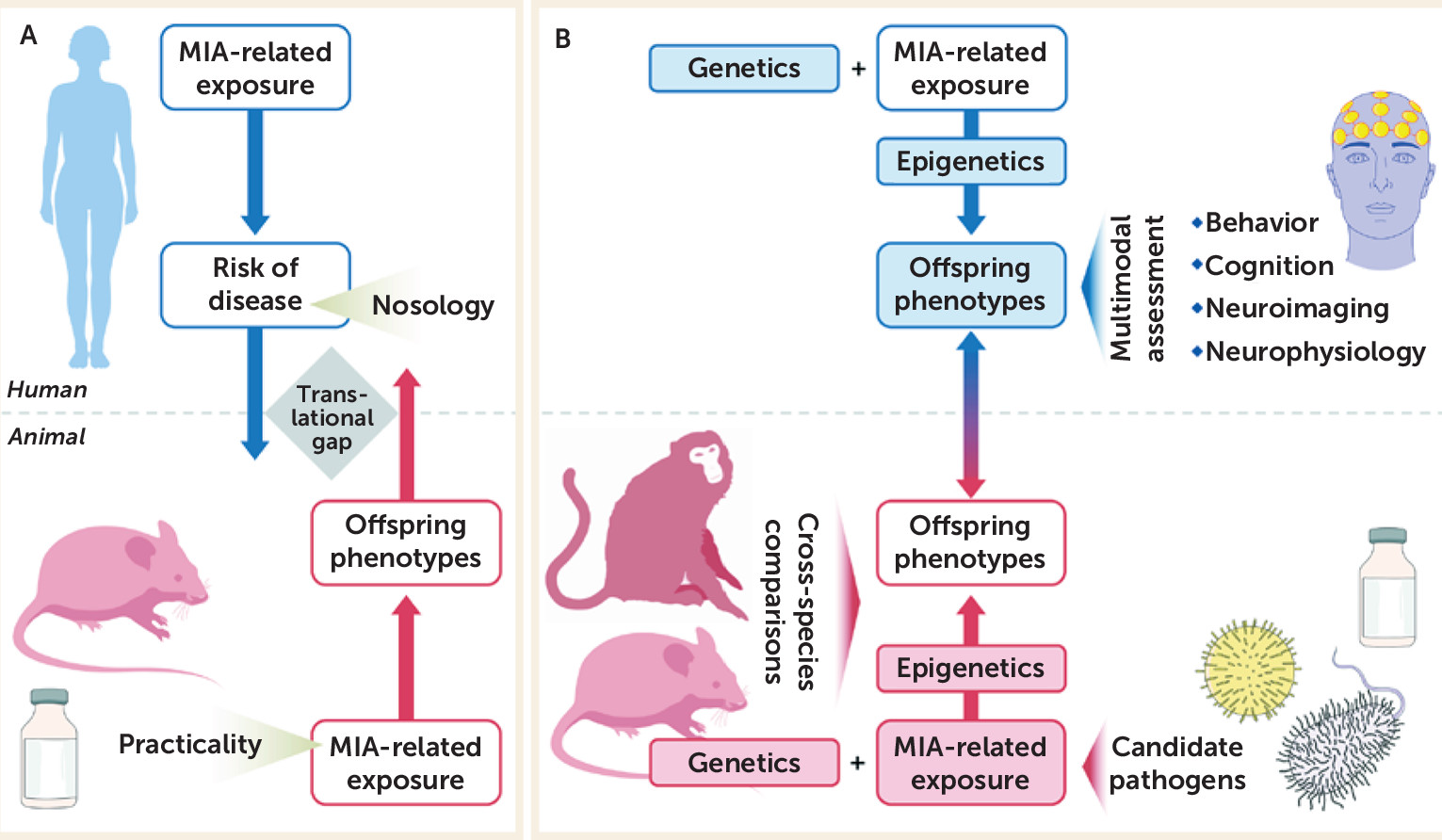
A key question is how the findings from the disciplines of epidemiology and basic science can complement and inform one another in furthering our understanding of the role of maternal immune activation in neuropsychiatric outcomes. In particular, we consider reverse translational approaches to this question, that is, whether human findings on maternal infection can predict parallel findings in experimental model systems. We first consider the parallels between the findings in epidemiologic and animal studies for schizophrenia, bipolar disorder, and autism spectrum disorder. Because a full review of the findings of maternal immune activation and neuropsychiatric outcomes is beyond the scope of this article, we highlight some key results and refer the reader to several comprehensive reviews (
1,
2,
6,
14–
18). Below, we focus on the potential areas of concordance between epidemiologic and basic science studies for each of these disorders.
Schizophrenia
To date, maternal immune activation and offspring psychiatric outcomes have been most commonly investigated for schizophrenia. We focus here on selected findings that are based on biomarkers of infection. Although not all findings have been replicated, key epidemiologic results include associations between maternal infectious pathogens (i.e., influenza virus, herpes simplex virus [HSV],
T. gondii, rubella, and bacterial pathogens) and inflammatory biomarkers (i.e., cytokines and C-reactive protein) and schizophrenia (
1,
59). Maternal exposure to influenza during early to mid-gestation, as quantified by antibody in maternal sera, was associated with a threefold increased risk of schizophrenia in the Child Health and Development Study (CHDS), which was based on a large birth cohort in northern California (
3). An elevated maternal
T. gondii IgG level was associated with a twofold elevation in schizophrenia risk in this same birth cohort (
60), and maternal genital and reproductive infections were associated with schizophrenia in this cohort (
61). Maternal exposure to HSV-2 was associated with nonaffective and affective psychoses in the National Collaborative Perinatal Project (
62) but not in the birth cohorts of the CHDS or of the Finnish Prenatal Studies, which is based on a large national birth cohort in Finland (
63). Neonatal antibodies to
T. gondii and cytomegalovirus have been associated with nonaffective psychosis in adulthood (
64). In our study of maternal cytokines in the CHDS, we observed that increased interleukin-8 was related to schizophrenia (
65). In the Finnish Prenatal Studies, we found that maternal C-reactive protein, a nonspecific biomarker of inflammation, was associated with an increased risk of schizophrenia (
59). Because it is unlikely that associations between biomarkers of inflammation are accounted for by one or a small group of infections, these findings may point to a common pathogenic pathway by which different infections give rise to schizophrenia.
Since their initial establishment, animal models of maternal immune activation have repeatedly documented structural and functional phenotypes that are implicated in schizophrenia and related psychotic disorders (
2,
14–
17). On the basis of early epidemiologic findings on maternal influenza and schizophrenia (
1), Fatemi et al. (
19–
22,
27) pioneered an experimental mouse model of prenatal exposure to human influenza virus in mice. As reviewed elsewhere (
14,
15), maternal influenza infection in mice led to a variety of behavioral, neurochemical, morphological, and transcriptional changes in the offspring, many of which are implicated in schizophrenia and related disorders. These findings are thus strongly related to, and provide experimental support for, the association between maternal influenza infection and risk of schizophrenia (
1,
3,
63). Since then, many additional investigations based on reverse translational animal models of maternal immune activation have yielded a wealth of new data supporting the predictive potential of epidemiologic studies. For example, deficits in sensorimotor gating, impairments in selective or sustained attention, deficiencies in working memory, and hyperresponsiveness to psychotomimetic drugs have been found in various rodent models of maternal immune activation, including prenatal exposure to influenza virus, the viral mimetic poly(I:C), the bacterial endotoxin lipopolysaccharide, and selected inflammatory cytokines (
14–
17). Some of these deficits show a maturational delay in their appearance and can be mitigated by symptomatic or preventive treatments with antipsychotic medications (
14–
17).
Notably, the fact that prenatal exposure to various immune-activating agents can elicit similar phenotypes is consistent with epidemiologic findings suggesting that the association between maternal immune activation and schizophrenia is not limited to a single infectious or inflammatory condition (
1,
15). Despite the similarities between maternal immune activation models, however, there are also some notable differences between the models with respect to the nature of brain and behavioral changes. For example, whereas prenatal poly(I:C) exposure in rats and mice has been shown to induce cellular, neurochemical, and behavioral phenotypes that are characteristic of a hyperdopaminergic state (
23,
66,
67), prenatal lipopolysaccharide exposure may instead induce a hypodopaminergic state in adult rodent offspring (
68). Prenatal lipopolysaccharide exposure in the rhesus monkey was found to cause a significant increase in global white matter volume (
50), whereas an opposite pattern (i.e., decreased white matter volume) was observed in rhesus monkey offspring born to mothers infected with influenza (
55). Besides the notable influence of prenatal timing and the genetic background discussed above, such differences may arise because different immunogens can induce a distinct set of neuroimmune abnormalities across brain development and, consequently, may lead to differing long-term deficits in brain structure and function. This notion would also be consistent with epidemiologic findings that appear to suggest that not all infectious pathogens have the same potential to increase neuropsychiatric disease risk (
1,
13). As discussed more extensively later, a closer examination of the commonalities and differences between the mediating factors and outcomes of distinct maternal immune activation models should help to further address this important issue.
Another question is whether animal models can also predict certain epidemiologic associations. Although comparatively little work has been done, our more recent findings support this assertion (
69). We developed an environmental two-hit model in mice, in which prenatal exposure to mild but physiologically relevant maternal immune activation served as the first hit, and subchronic exposure to unpredictable psychological stressors in pubescence served as the second hit (
69). Hence, this multifactorial model incorporates two environmental risk factors that have each been associated with increased risk of psychiatric disorders such as schizophrenia. We showed that combined exposure to the two environmental adversities acted in synergy to induce psychosis-related neural and behavioral abnormalities in adult mice (
69). These results provided the first evidence, to our knowledge, suggesting that prenatal immune adversities can function as a neurodevelopmental disease primer, which in turn can increase the offspring’s vulnerability to the detrimental neuropathological effects of subsequent stress exposure during pubescence (
69). These basic science findings have recently been translated to a large population-based epidemiologic study, which comprised nearly 1 million Danish persons born between 1980 and 1998 (
70). In that study, Danish nationwide registers were linked to estimate the independent and joint effects of exposure to prenatal infection and peripubertal psychological trauma on the risk of schizophrenia. Prenatal exposure to infection was defined on the basis of records of hospital admissions with an infection during pregnancy, and exposure to traumatizing experiences during peripuberty (ages 8 to 14 years) was defined according to Danish standards and included parental death, maltreatment or physical or sexual abuse, and maternal and paternal occupational situation and history of crime. Confirming the hypothesis initially put forward by the environmental two-hit model in mice (
69), the Danish study demonstrated that exposure to prenatal infection and peripubertal psychological trauma was associated with a significantly higher risk of developing schizophrenia (among males) compared with exposure to either insult alone, and the interaction between infection and trauma attained statistical significance (
70). These findings suggest that the cross-fertilization between basic research in animals and risk factor epidemiology may offer the potential for predicting yet undiscovered associations between maternal immune activation and neuropsychiatric illnesses.
Bipolar Disorder
To date, only a few epidemiologic studies have evaluated maternal immune activation associated with bipolar disorder in offspring. Our group has demonstrated that maternal influenza, documented by antibodies in prenatal sera (
59) and physician diagnoses (
71), is associated with a fivefold increased risk of bipolar disorder. Although most other studies suggest no association between maternal infectious pathogens and bipolar disorder (
13), one study found that maternal exposure to the type 1 strain of
T. gondii was related to an increased risk of affective psychoses in offspring, which includes bipolar disorder (
72).
Even though animal models of maternal immune activation have not been specifically explored for their validity for bipolar disorder, some of the experimentally induced phenotypes may be relevant for this neuropsychiatric illness as well. For example, deficits in sensorimotor gating, as seen in various rodent maternal immune activation models (
2,
14–
17), are also present in patients with acutely manic (
73) and remitted bipolar disorder (
74). Moreover, several animal studies have reported the emergence of depression-like behaviors in offspring exposed to maternal immune activation (
75,
76). The latter phenotypes may be relevant not only for unipolar depression but also for depressive episodes in bipolar disorder. The investigation of other core behavioral symptoms of bipolar disorder, such as poor decision making, altered risk-taking behavior, impulsivity, and loss of inhibitory control, remain unexplored in maternal immune activation models. Additional work is necessary to evaluate whether maternal immune activation–induced deficits can be mitigated by pharmacological treatments used in bipolar disorder, including the mood stabilizer lithium and anticonvulsants such as valproate and lamotrigine (
77).
Autism
In recent years, maternal infection and inflammation have been investigated in relation to autism spectrum disorder. Although findings are mixed and more work is necessary, evidence has emerged linking maternal inflammation to the risk of autism spectrum disorder in offspring. In the Finnish Prenatal Studies birth cohort, our group demonstrated that elevated maternal levels of C-reactive protein, a nonspecific biomarker of inflammation, in early to mid-gestation was related to an increased risk of autism spectrum disorder in offspring (
4). However, in the Early Markers for Autism study conducted in northern California, maternal mid-pregnancy levels of C-reactive protein were related to a decreased risk of autism spectrum disorder (
78). Examination of cytokines and chemokines in archived maternal serum samples in the Early Markers for Autism study demonstrated that significantly increased levels of these analytes were related to autism spectrum disorder (
5,
11). In amniotic fluid samples from a Danish study, several cytokines, including tumor necrosis factor-alpha, and several inflammatory interleukins were related to autism spectrum disorder in offspring (
7). Moreover, maternal fever has been associated with autism. Although replication of these findings is necessary, they suggest that maternal immune activation may be related to autism spectrum disorder. Consistent with this interpretation, other maternal immune factors, including maternal autoantibodies targeting fetal proteins, have been associated with increased risk of autism spectrum disorder in offspring (for a review, see reference
79). These findings include significant associations between paired maternal antibody reactivity to fetal brain proteins with the 37-kDa and 73-kDa molecular weight bands and diagnosis of autism spectrum disorder in children (
80). Within proteins corresponding to the 37-kDa, 39-kDa, and 73-kDa bands, maternal autoantibodies recognized seven developmentally regulated proteins in the fetal brain, including lactate dehydrogenase A and B, stress-induced phosphoprotein 1, and collapsin response mediator proteins 1 and 2 (
79,
80). Several of these proteins are critical for normal brain development, including neuronal migration and neural network formation.
Animal models further support the hypothesis that maternal immune activation is an environmental risk factor for autism spectrum disorder. For example, prenatal exposure to the viral mimetic poly(I:C), the bacterial endotoxin lipopolysaccharide, or allergies and asthma can all induce behavioral abnormalities that are reminiscent of core symptoms of autism spectrum disorder, including deficits in social interaction and communication and high levels of repetitive behaviors (
9,
15,
34,
81). These manipulations also cause brain morphological and cellular abnormalities implicated in autism spectrum disorder, including abnormal cerebellar development, impaired expression of the extracellular matrix protein reelin, and altered synapse density and neural connectivity (
2,
15,
21). Notably, some of these rodent findings have been extended to rhesus monkeys, both at the behavioral and brain morphological levels (
8,
50,
56,
82).