Identification of brain network substrates of the symptoms of psychiatric illnesses has been a primary goal of psychiatric neuroimaging. Numerous factors have confounded the discovery of brain circuits and network dysfunction that cause symptoms and disability. Diagnostic heterogeneity, illness chronicity, technical limitations, and methodological differences have limited our ability to demonstrably identify the network basis of disabling psychiatric symptoms (
1).
Psychotic disorders such as schizophrenia are disabling lifelong illnesses that afflict more than 3 million people in the United States. A robust literature has examined how psychotic symptoms predict functional outcomes such as employment, social functioning, and independent housing. These studies have demonstrated that the most readily recognizable positive symptoms, such as delusional thought and hallucinations, are not the best predictor of the functional status of individuals with psychosis. Rather, it is the severity of negative symptoms, such as amotivation, expressive deficits, and anhedonia, that best predicts functional outcomes and overall quality of life (
2,
3). The development of new interventions for these negative symptoms is hampered by a critical shortfall in our understanding of their pathophysiology, and our ability to modify the functional impact of schizophrenia is further limited by the fact that psychotropic medications have limited efficacy in treating negative symptoms (
4,
5).
Neuroimaging has the potential to illuminate the neuroanatomical basis of these deficits, but existing studies have not converged on a consensus circuit or network target for amelioration in the clinic. Potential explanations for this nonconvergence include methodological heterogeneity between studies (
6), the effects of individual variance within a diagnosis (e.g., duration of illness [
7]), and the possibility that multiple network pathology subtypes may present with similar clinical features (as suggested by Drysdale et al. [
8] for depression). Even in scenarios in which there is agreement between studies, most imaging studies are inherently correlational and cannot establish a causal relationship between imaging signal and the phenotype of interest—that is, these findings may be causal to clinical manifestations of the illness, functionally compensatory processes, or purely epiphenomena. To address these concerns, there is a growing consensus that symptom-pathophysiology relationships may be best tested using a within-subjects design in which pathophysiology is experimentally manipulated to determine the impact on symptoms (
9).
We therefore propose a strategy wherein a symptom-related circuit is empirically discovered, and then, in a separate cohort, this circuit is challenged in a targeted way to assess causal relations (
10). Recent advances in noninvasive brain stimulation have demonstrated the feasibility of manipulating single-network connectivity (
11) in a way that uniquely affects the targeted network (
12) and concurrently affects behavior (
13). This hypothesis predicts that if circuit pathology and psychiatric symptoms are causally linked, then it must follow that the manipulation of the circuit should be reflected in symptom modification.
In a cohort of participants with early-course schizophrenia or schizoaffective disorder, we sought to identify brain network correlates of negative symptoms. Methodological decisions included recruitment of participants with early-course illness to mitigate the effects of illness chronicity, imaging processing tailored to mitigate movement effects, and analysis using an unbiased, data-driven approach to locate symptom-imaging correlates at the individual voxel level. After identifying this network, we examined, in a separate experimental cohort, the effect of repetitive brain stimulation at the network node most strongly associated with symptom severity. We show that a frontal-cerebellar circuit abnormality is associated with negative symptoms and that noninvasive brain stimulation that modulates this circuit abnormality ameliorates negative symptoms, thus revealing a causal relation between the brain circuit abnormality and the clinical symptoms.
Methods
Participants
Network discovery.
Data on 44 participants with schizophrenia (N=35) or schizoaffective disorder (N=9) who were recruited for a clinical trial (NCT01561859) were included in the study. Before participation in the study, all participants provided written informed consent in accordance with the institutional review boards of the University of Pittsburgh and Beth Israel Deaconess Medical Center (Boston).
Network validation.
Data on 11 participants with schizophrenia recruited for a clinical trial (NCT01551979) were also included in the study. All participants provided written informed consent in accordance with the institutional review board of the Beth Israel Deaconess Medical Center.
MRI Acquisition
Imaging was conducted on Siemens 3.0-T MRI systems (Munich, Germany). Briefly, 1-mm3 T1-weighted anatomical scans were acquired, and multiple functional runs of approximately 6 minutes were acquired from all participants (124 time points, 3-second repetition time, 3-mm3 voxels).
MRI Data Processing
All analyses were preprocessed using the DPABI toolbox (Data Processing and Analysis for Brain Imaging) (
14;
http://rfmri.org/dpabi). As a quality control metric, scans that exceeded motion thresholds (>3 mm translation or >3° rotation) were discarded. Individual time points with framewise displacement >0.5 mm were discarded, and scans with >50% of volumes removed for framewise displacement were discarded. All data were preprocessed to remove motion (24-parameter), CSF signals, white matter signals, and an overall linear trend. A bandpass filter was applied (0.01–0.08 Hz). Data were normalized using the DARTEL toolbox (
http://www.neurometrika.org/node/34) into Montreal Neurological Institute (MNI) space and smoothed with an 8-mm full-width half-maximum kernel. Analyses were conducted in a gray matter mask defined within the group.
Network identification was conducted with multivariate distance matrix regression. Time courses from regions identified with the network identification method were extracted using the DPABI toolbox for the network validation cohort and then correlated with z-transformed Pearson’s correlation coefficients. An additional analysis was conducted with SPM12 for voxel-wise maps.
Clinical Assessment
Participants were assessed with the Scale for Assessment of Negative Symptoms in the network discovery cohort and with the Positive and Negative Syndrome Scale (PANSS) for the network validation cohort.
Additional methodological details are presented in the online supplement.
Results
Network Discovery With Multivariate Distance Matrix Regression (MDMR)
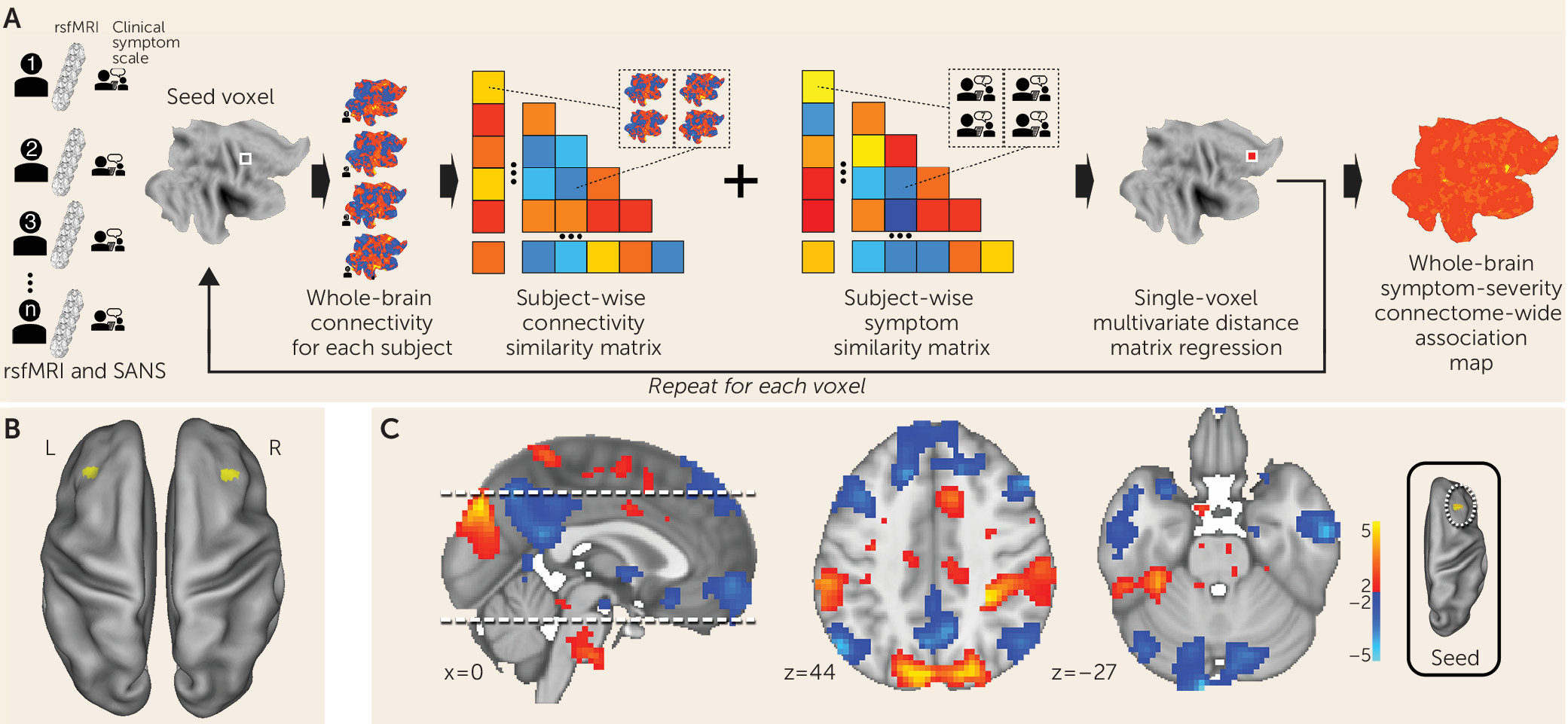
MDMR reveals functional connectivity correlates of negative symptoms in schizophrenia.
We examined clinical, demographic, and resting-state functional MRI (fMRI) data on 44 participants diagnosed with schizophrenia or schizoaffective disorder (
Table 1) (for further details on network discovery, see the
online supplement). Negative symptom severity was quantified by trained raters who administered the Scale for the Assessment of Negative Symptoms (
15). For preprocessing resting-state fMRI data, we used procedures optimized to control for motion-related effects (
16). Individual fMRI scans were coregistered into a common space (MNI). After preprocessing, resting-state fMRI data were analyzed with MDMR (
Figure 1A) (
17). This method allows for an unbiased, data-driven approach to determining functional connectivity. In contrast to most other approaches, MDMR allows quantification of how a variable of interest (negative symptom severity here) is reflected in the connectivity of individual voxels to the whole brain (i.e., at the finest resolution possible) without parcellating the brain into regions defined a priori (
Figure 1A) (for further details, see the
online supplement). This approach has been used to examine the relationship between psychiatric pathology and connectivity (
18–
21). We modeled the effect of negative symptom severity on functional connectivity while covarying for effects of in-scanner motion, age, sex, study site, and prescribed medication dosage. This analysis identified the middle frontal gyrus (Brodmann’s area 9) in the dorsolateral prefrontal cortex bilaterally as the regions where functional connectivity covaried significantly with negative symptom severity. The right dorsolateral prefrontal cortex (peak voxel z=3.8; MNI coordinates: 36, 24, 30) demonstrated a more significant relationship between functional connectivity and negative symptom severity than the left dorsolateral prefrontal cortex (peak voxel z=2.95; MNI coordinates: −33, 30, 42) (
Figure 1B).
Negative symptom severity is inversely correlated with functional connectivity between the right dorsolateral prefrontal cortex and the default mode network.
MDMR identifies brain regions where connectivity shows a significant association with a phenotypic variable of interest. Post hoc testing is then necessary to determine the spatial distribution and the directionality of the connectivity. We calculated the subject-specific functional connectivity in the right dorsolateral prefrontal cortex and correlated these maps with negative symptom severity to identify the network that differentially connects to this region depending on symptom severity. We observed that negative symptom severity was inversely correlated with right dorsolateral prefrontal cortex connectivity to a distributed brain network that includes both cerebral and cerebellar nodes of the default network (
22,
23) (
Figure 1C). Notably, we found that a connectivity breakdown between the right dorsolateral prefrontal cortex and the midline cerebellar node in the default network (MNI coordinates: −9, –96, −27) was the most significant predictor of negative symptom severity.
In summary, we used a purely data-driven analysis to identify the most significant functional connectivity correlate of negative symptom severity in a sample of individuals with schizophrenia or schizoaffective disorder. If disconnectivity between the right dorsolateral prefrontal cortex and the cerebellum is causally related to negative symptom severity, then selectively reversing disconnectivity should be reflected in a reduction in negative symptom severity. We therefore sought to validate or refute a causal relationship between network disconnectivity and negative symptoms.
Network Validation With Transcranial Magnetic Stimulation (TMS)
Empirically testing the causal relationship between network connectivity and negative symptom severity.
We and other investigators have previously demonstrated that repetitive TMS (rTMS) can selectively modulate network functional connectivity in healthy individuals (
11,
12,
24). We hypothesized that if breakdown in dorsolateral prefrontal cortex connectivity is causally linked to negative symptom severity, rTMS restoration of functional connectivity should be reflected in amelioration of negative symptom severity.
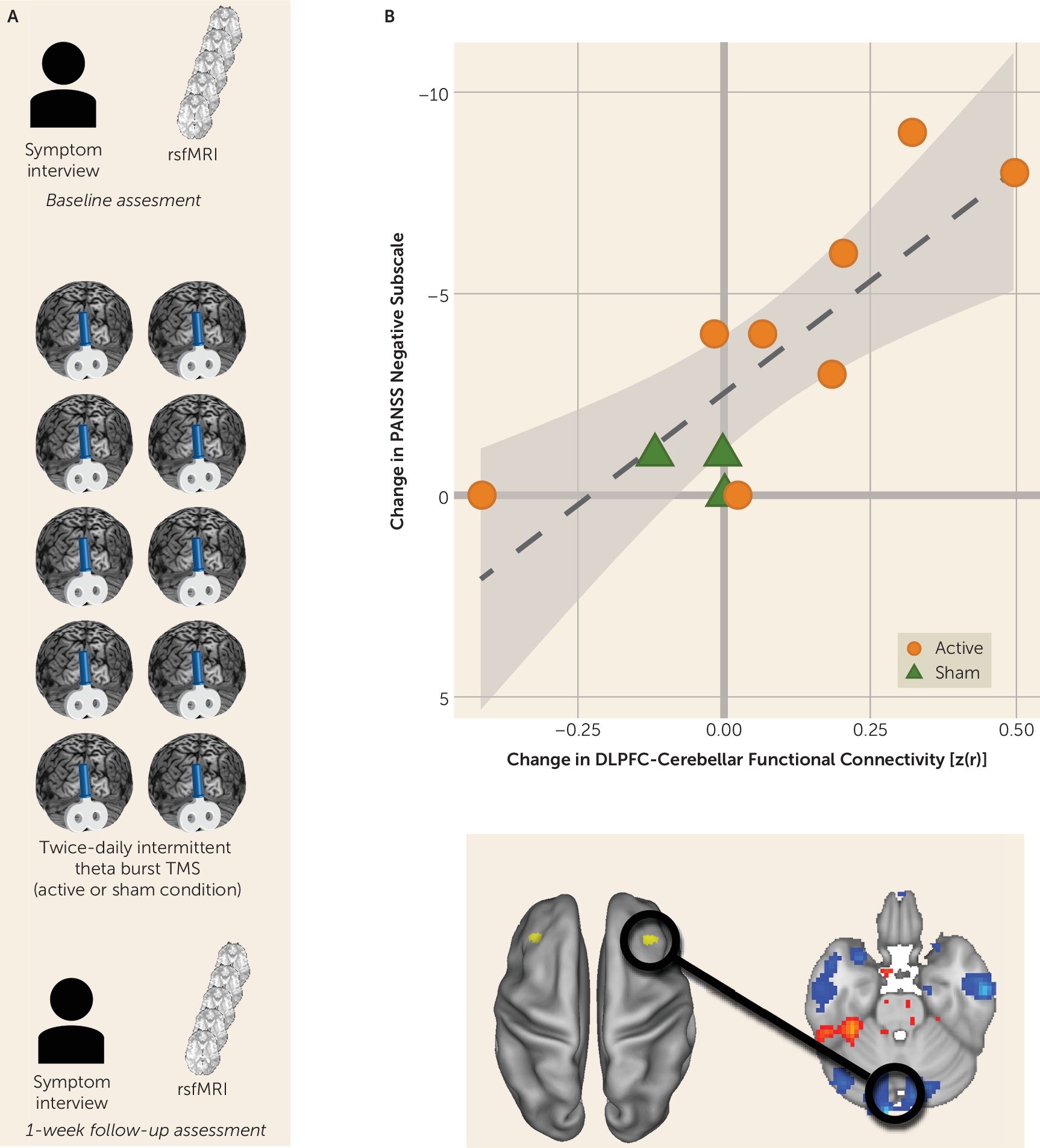
The most significant relationship between disconnectivity and symptom severity was between the right dorsolateral prefrontal cortex and the midline cerebellum. We therefore examined an independent, testing cohort of participants with schizophrenia who underwent an interventional, sham-controlled trial of rTMS that targeted the cerebellar vermis (midline) (N=11) (
Figure 2A; for further details, see the
online supplement). Participants underwent clinical characterization by a trained rater who was blind to the treatment condition. Negative symptom severity was quantified by using the negative symptom subscore from PANSS (
25). Participants underwent a baseline resting-state fMRI scan and then were randomly assigned to either active or sham rTMS modulation of the cerebellum. Intermittent theta burst (10 bursts of three biphasic pulses at 50 Hz, repeated at 5 Hz, for 10 seconds for a total of 600 pulses) of 100% of active motor threshold was applied to the midline cerebellum. Sham electrodes were placed at the neckline on all study subjects. Blinding codes were used to determine which side of an active or passive Magpro coil (MagVenture Cool B65 A/P, MagVenture A/S, Farum, Denmark) was used for stimulation.
Participants underwent two rTMS sessions per day separated by 4 hours for 5 days. This intensity, frequency, and duration of stimulation was chosen on the basis of demonstrated efficacy in ameliorating negative symptoms at the group level (
26). After the week of repetitive stimulation, participants underwent clinical characterization and a follow-up resting-state fMRI scan (
Figure 2A). We hypothesized that rTMS-induced change in functional connectivity and change in negative symptom severity would be correlated for all participants (i.e., regardless of whether they received sham or real stimulation). Specifically, an increase in cerebellar-dorsolateral prefrontal cortex connectivity should be reflected by a reduction in symptom severity.
Reversing cerebellar-dorsolateral prefrontal cortex disconnectivity ameliorates negative symptom severity.
We measured pretrial-to-posttrial within-subject change in negative symptom severity as well as change in functional connectivity between the right dorsolateral prefrontal cortex and the cerebellar node of the default network. As predicted, we observed a strong and significant relationship between increased connectivity and reduction in symptom severity (r=−0.809, p=0.003; 95% CI=−0.948, −0.405) (
Figure 2B).
Testing distributed-network compared with cerebellar-specific effects on negative symptoms.
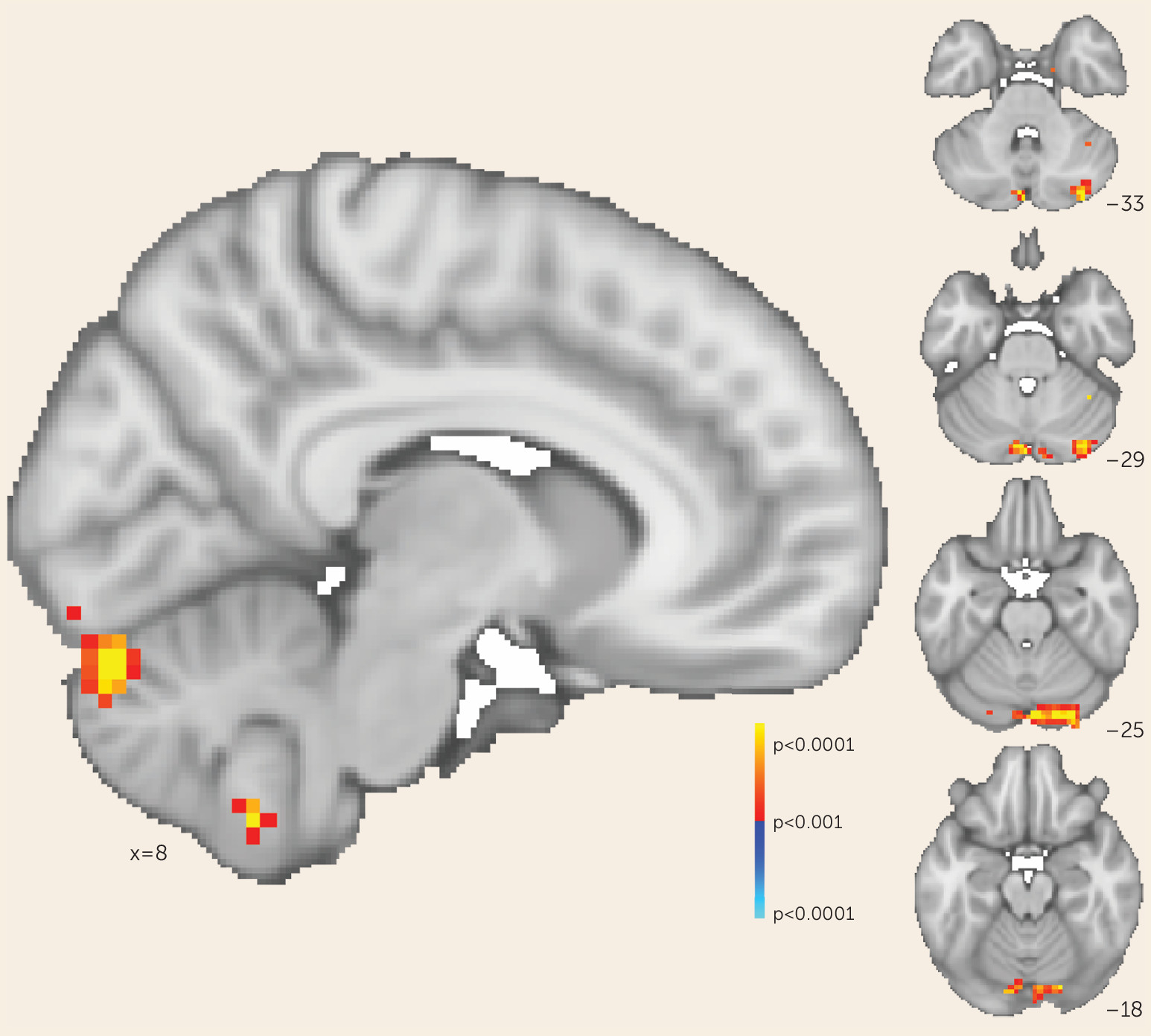
Consistent with our original hypothesis, we observed that selectively increasing functional connectivity between the cerebellar node of the default network and the right dorsolateral prefrontal cortex resulted in reduced negative symptom severity. We sought to determine whether this effect was mediated solely by changes in cerebellar-dorsolateral prefrontal cortex connectivity or was the result of cerebellar rTMS increasing functional connectivity broadly between the right dorsolateral prefrontal cortex and the rest of the default mode network—that is, both cerebellar and cerebral nodes. We tested these possibilities by generating whole-brain maps of change in functional connectivity (pre-rTMS compared with post-rTMS) to the right dorsolateral prefrontal cortex for each participant. We then regressed these maps against individual change in negative symptom scores to determine where connectivity change correlated with symptom change. This whole-brain analysis identified one area where connectivity change corresponded to symptomatic change: the cerebellum at the level of rTMS stimulation (p<0.001). In this region, the correlation between symptomatic improvement and connectivity change was particularly strong (r=−0.952, 95% CI=−0.987, −0.821). The cerebral nodes of the default network were not identified by this analysis even at a lower threshold (p<0.05) (
Figure 3). This suggests that in our trial, symptomatic improvement across all participants was mediated by rescue of cerebellar-dorsolateral prefrontal cortex functional connectivity rather than a broader modulation of multiple cerebral networks, as would be expected if this network connectivity mediated negative symptoms.
We also found that the active stimulation condition was significantly more effective than the sham condition in increasing cerebellar-dorsolateral prefrontal cortex connectivity (t=2.938, df=8.96, p=0.017) and in reducing negative symptom severity (t=2.931, df=7.99, p=0.019).
In summary, we used a combination of resting-state fMRI and multivariate data analysis to discover a dorsolateral prefrontal cortex-default network connectivity breakdown associated with schizophrenia negative symptom severity. This connectivity breakdown was strongest between the dorsolateral prefrontal cortex and the midline cerebellum. When repeated rTMS stimulation was used to selectively rescue cerebellar-dorsolateral prefrontal cortex connectivity, negative symptom severity was ameliorated.
Discussion
The ability to use biology to differentiate phenotype and prognosis has improved greatly in recent years, but most studies to date have not been structured to answer questions about the causal relationship between biomarker and behavioral phenotype. This inability to differentiate the biology that mediates disease from compensatory processes or obligatory epiphenomena has led to a “causality gap in human psychiatric neuroscience” (
27). In addition to limiting our understanding of the basic neuroscience of psychiatric illness, the absence of a biological target that mediates disease state means that even large trials that evaluate symptomatic response to an intervention cannot lead to a greater understanding of why some patients respond and others do not.
In this study, we combined two mature technologies for neuroscience discovery (resting-state fMRI and rTMS) and applied them to better understand disabling, medication-refractory symptoms in schizophrenia. Using recent developments in data-driven imaging data analysis, we identified a network biomarker of negative symptom severity in a sample of patients with schizophrenia or schizoaffective disorder. We tested the causal nature of network dysfunction through the selective modulation of this network in a trial designed to hold other factors constant (i.e., a within-subject design). We observed that changing cerebellar-dorsolateral prefrontal cortex connectivity appeared to reverse the experiential and expressive deficits referred to as negative symptoms. This finding provides empirical support for a causal relationship between dysfunctional connectivity and psychopathology.
Our findings suggest the existence of at least one network circuit linked directly to negative symptoms. Investigators using alternative approaches have identified different network-symptom relationships (
28–
30). We suspect that the network we identified is present within these previous data sets; however, it may be obfuscated by common confounders in psychiatric neuroimaging, such as diagnostic heterogeneity, duration of illness, and technical considerations, including motion in the scanner. Although our network discovery data set consisted of patients with early-course schizophrenia (<8 years since their diagnosis), we were able to validate this symptom-connectivity relationship even in participants with later-course illness. This suggests that there is a consistent network-symptom relationship that may be inconsistently observed in purely imaging studies.
There are some existing preliminary hypotheses regarding the emergence of negative symptoms from cerebellar-dorsolateral prefrontal cortex connectivity. Previous imaging studies have identified frontal abnormalities that may be associated with negative symptoms in schizophrenia patients (
31,
32). What is the significance of identifying the dorsolateral prefrontal cortex as one node of a circuit that includes the cerebellum? Our findings may be seen as consistent with a dysmetria of thought theory (
33–
35) that hypothesizes that just as the cerebellum regulates the rate, rhythm, force, and accuracy of movements, so does it regulate the speed, consistency, capacity, and appropriateness of mental or cognitive processes, including those subserved by the prefrontal cortex. It has been hypothesized that cerebellar dysfunction may underlie the pathophysiology of schizophrenia (
36–
41), but this has not been demonstrated until now.
In sum, the precise localization of a prefrontal-cerebellar network is critical in identifying the underlying neural substrates of the disabling negative symptoms in schizophrenia. Our results provide experimental evidence in support of such a circuit, which, when directly modulated, rescues these deficits.
Our results are consistent with a potentially reproducible model of TMS-based rescue of a breakdown of connectivity. Two previous studies demonstrated a similar reduction in negative symptom severity at the group level after 10 sessions of intermittent theta-burst stimulation to the cerebellar vermis (
26,
42). Unfortunately, although these studies produced potential therapeutic improvement, they provided no mechanistic explanation of how improvement occurred, nor did they demonstrate that improvement was a result of correction of existing circuit pathology. Taken together with the present data, these findings suggest an effect that is readily replicable. Furthermore, our network identification and TMS samples were collected at different facilities, which strengthens the generalizability of our results.
Our study has several strengths. First, it establishes a causal relationship between resting-state functional connectivity and disease expression in a psychiatric illness, moving the field away from purely correlational studies. Second, it establishes functional connectivity between the dorsolateral prefrontal cortex and the cerebellum as a quantifiable and engageable target that modulates disabling, medication-refractory negative symptoms in schizophrenia. Third, it is a model of how a precision-medicine approach (i.e., targeting disease-specific pathophysiology to change clinically observable symptoms) may be applied to psychiatric disorders. Fourth, by linking neuromodulation to a biological outcome (functional connectivity) rather than to symptomatic response alone, this model allows individual-level explanation of response and nonresponse to rTMS.
Limitations of this study include the small sample size of the network validation (TMS) experiment. In a traditional rTMS clinical trial with clinical response as the only readout of TMS engagement, our sample size would be inadequate to draw conclusions about the cerebellum’s role in negative symptoms. By combining clinical response with functional connectivity, our study was more than sufficiently powered to discover the strong relationships between connectivity and symptoms. However, with a larger sample size, we could have addressed questions about what factors mediate connectivity change (and therefore symptom change) in response to rTMS—for example, Do individual differences in duration of illness and network topography modulate connectivity change to a course of rTMS? Additionally, we did not assess participant and rater blinding, a procedure that may be helpful in the assessment of placebo response in future studies.
Finally, while our findings demonstrate a target biological substrate for the treatment of negative symptoms, we do not suggest that cerebellar-targeted TMS is the sole intervention that can do so. Our ability to measure cerebellar-dorsolateral prefrontal cortex circuit integrity by using fMRI suggests that functional connectivity may be a useful marker of efficacy for other therapeutic interventions in the treatment of negative symptoms, independent of rater assessment. Similarly, previous TMS studies did not systematically investigate stimulation parameters, and it is therefore plausible that our connectivity assessment may be useful as a direct measure of rapid protocol development.
Acknowledgments
The authors thank Zarrar Shehzad and Neeraj Tandon for their assistance with multivariate distance matrix regression analysis; Faranak Farzan, Andrea Pousada, Allison Glasgow, Alice Lee, Cristina Torres-Rivas, Bonnie Wong, Mark Eldaief, Adam Stern, and Ann Connor as well as the staff of the Beth Israel Deaconess Medical Center-Harvard Catalyst and of the Harvard Center for Brain Science Neuroimaging facility for their assistance in the collection of the transcranial magnetic stimulation trial data; Himanshu Bhat and Thomas Benner of Siemens Healthcare for the simultaneous multislice-echo planar imaging sequence; Steven Cauley of Massachusetts General Hospital for modifications that enabled implementation of MRI protocols; and the patients of the Brain Imaging, Cognitive Enhancement and Early Schizophrenia and transcranial magnetic stimulation trials.