Methods
Institutional review boards (in the United States) or independent ethics committees (in Europe) at the various study sites approved the study protocol and amendments. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practices, and applicable regulatory requirements. All patients provided written informed consent before participating in the study.
Study Design
This was a phase 3 randomized, double-blind, active-controlled, multicenter study conducted between August 2015 and November 2017 at six sites in the Czech Republic, nine in Germany, seven in Poland, seven in Spain, and 10 in the United States.
The study consisted of three phases: 1) a 4-week screening and prospective observation phase during which treatment response to the current ongoing oral antidepressants was assessed; 2) a 4-week treatment phase during which participants received a new oral antidepressant combined with either esketamine nasal spray or placebo nasal spray; and 3) a posttreatment follow-up phase of up to 24 weeks. At study entry, participants had documented (retrospectively) nonresponse (≤25% improvement) to one to five antidepressants (based on the Massachusetts General Hospital [MGH] Antidepressant Treatment Response Questionnaire [
13]) in the current depressive episode and were currently receiving a different oral antidepressant, to which they had been adherent (<4 missed days of treatment based on the Patient Adherence Questionnaire) for at least the previous 2 weeks at or above the minimum therapeutic dosage (or therapeutic blood level for specific tricyclic antidepressants), which was continued prospectively for another 4 weeks during the screening/prospective observational phase, providing a total duration of at least 6 weeks of the prospective antidepressant.
To determine patient eligibility, an independent clinical reviewer (a psychiatrist or psychologist) confirmed the adequacy of the prospective antidepressant medication and evaluated the patient’s ratings on the Montgomery-Åsberg Depression Rating Scale (MADRS) (
14), the Mini International Neuropsychiatric Interview (
15), the MGH Antidepressant Treatment Response Questionnaire, the Columbia-Suicide Severity Rating Scale (C-SSRS) (
16), the Inventory of Depressive Symptomatology–Clinician Rating (IDS-C) (
17), and the Clinical Global Impressions severity scale (CGI-S) (
18) at screening, as well as medical history and concomitant therapies.
Patients who had not responded to the prospective antidepressant treatment by the end of the screening phase (nonresponse was defined as ≤25% improvement in MADRS score from week 1 to week 4 and a MADRS score ≥28 at weeks 2 and 4) entered the 4-week double-blind treatment phase, at which time they discontinued all current antidepressant treatments and were randomly assigned to the intranasal treatment combined with a newly initiated oral antidepressant.
Study Population
To be eligible, participants had to be between 18 and 64 years of age; have either single-episode (≥2 years) or recurrent major depressive disorder (per DSM-5 criteria) without psychotic features, confirmed by the Mini International Neuropsychiatric Interview; have a score ≥34 on the IDS-C, consistent with moderate to severe depression; and meet the study definition of treatment-resistant depression, which was nonresponse to an adequate trial (dosage, duration, and adherence) of at least two antidepressants in the current episode (of which one was observed prospectively). Patients also had to be medically stable (those on thyroid hormones had received a stable dosage for ≥3 months) and able to self-administer intranasal medication.
Key exclusion criteria were current or recent (past 6 months) homicidal ideation/intent or suicidal ideation with intent to act or suicidal behavior within the past year; diagnosis of psychotic disorder, major depressive disorder with psychotic features, bipolar or related disorders, borderline, antisocial, histrionic, or narcissistic personality disorder, obsessive-compulsive disorder (current), intellectual disability, autism spectrum disorder; uncontrolled hypertension; seizures; a recent history (past 6 months) of moderate or severe substance use disorder (including a lifetime history of ketamine use disorder); and positive urine test results for specified drugs of abuse (cannabinoids, barbiturates, methadone, opioids, cocaine, phencyclidine, and amphetamine/methamphetamine). A urine drug screen with adequate assay sensitivity and specificity for ketamine and derivatives was not available and therefore was not performed. Patients who had previously shown nonresponse to esketamine or ketamine, nonresponse to the oral antidepressant options for this study (detailed below), or nonresponse to electroconvulsive therapy were excluded. Participants were required to use adequate contraception during the study. A full list of the inclusion and exclusion criteria is presented in the online supplement.
Randomization and Blinding
A computer-generated randomization schedule was used to randomly assign eligible patients in a 1:1 ratio to receive double-blind treatment with either esketamine (56 mg or 84 mg) nasal spray (hereafter referred to as esketamine) or placebo nasal spray (hereafter referred to as placebo), administered twice weekly, each combined with a newly initiated open-label oral antidepressant administered daily. Randomization was balanced by using randomly permuted blocks and was stratified by country and by class of oral antidepressant (serotonin-norepinephrine reuptake inhibitor [SNRI] or selective serotonin reuptake inhibitor [SSRI]). Patients, investigators, site personnel, those assessing outcomes, and those analyzing the data were blind to treatment assignment.
Intranasal Study Drug and Administration
Both intranasal study drugs (esketamine and placebo) were provided in disposable nasal spray devices with identical appearance and packaging. Each device contained 200 μL of solution and delivered two sprays of either esketamine (for a total dose of 28 mg per device) or placebo. To maintain blinding, a bittering agent was added to the intranasal placebo to simulate the taste of the esketamine solution, and three devices were administered to all patients at all sessions; in the esketamine plus antidepressant arm, a total dose of 56 mg was delivered by use of two active devices and one placebo device, and a total dose of 84 mg was delivered by use of three active devices (see Table S2 in the online supplement).
All participants received training and practiced using the intranasal device before the first administration. Participants self-administered intranasal study drug at the clinical site under the direct supervision of the investigator.
For improved tolerability, esketamine was started on day 1 at 56 mg (see Table S3 in the online supplement), with the possibility, per the investigator’s clinical judgment based on efficacy and tolerability, of increasing the dose to 84 mg or remaining at 56 mg on days 4, 8, 11, or 15 (after which the dose remained stable).
Newly Initiated Oral Antidepressant
The open-label oral antidepressant initiated with the intranasal study drug was selected by the investigator from two different drug classes: an SSRI (escitalopram or sertraline) or an SNRI (duloxetine or venlafaxine extended release), provided by the sponsor. The investigator chose one of the four options (representing the most commonly used standard-of-care antidepressants) based on review of the patient’s MGH Antidepressant Treatment Response Questionnaire and relevant prior antidepressant medication information, and considering that the selected antidepressant was one with which the patient had not previously experienced nonresponse (in the current depressive episode) or been intolerant to (lifetime), and was available in the participating country.
Dosing of the oral antidepressant followed a fixed titration schedule (presented in Table S4 in the online supplement).
Efficacy Assessments
Because esketamine exhibits transient dissociative effects that are difficult to blind, possibly biasing the site staff supervising the dosing, all MADRS assessments (used for the primary endpoint, the first key secondary endpoint, and calculation of response and remission rates) were performed by independent, remote (by telephone) raters who were blind to the protocol details, including study visit, the patient’s clinical status, and side effects during the trial.
Patients rated the impact of the study treatments on socio-occupational disability using the Sheehan Disability Scale (
19), on depressive symptoms using the 9-item Patient Health Questionnaire (PHQ-9) (
20), on severity of anxiety using the 7-item Generalized Anxiety Disorder scale (
21), and on overall health outcome using the EuroQol-5 dimension-5 level (EQ-5D-5L [
22]; results reported in the
online supplement) at baseline and on days 15 (except the Generalized Anxiety Disorder scale) and 28. Investigators rated change in severity of depressive illness using the CGI-S.
Safety Assessments
Adverse events and other safety assessments (hematology and serum chemistry, urinalysis, physical examination, electrocardiogram, C-SSRS) were monitored throughout the study. Vital signs, the Clinician-Administered Dissociative States Scale (CADSS) (
23), and the 4-item positive symptom subscale of the Brief Psychiatric Rating Scale (BPRS) (
24) were assessed at baseline and at all dosing visits (before dosing and at 40 minutes, 1 hour [vital signs only], and 1.5 hours after dosing). Investigators were provided guidance on blood pressure monitoring on intranasal treatment days (see the
online supplement). Investigators were also instructed on being watchful of requests for an increase in intranasal study drug dose and/or dosing frequency, which may indicate early signs of abuse or addiction.
The Modified Observer’s Assessment of Alertness/Sedation scale (scored from 0 [no response to painful stimuli] to 5 [readily responds to name spoken in normal tone]) was used to assess the level of postdose sedation every 15 minutes from before dosing to 1.5 hours after dosing.
Investigators assessed patients’ clinical status and discharge readiness (based on their overall clinical status, including sedation, blood pressure, and other adverse events) using a scale developed for the program—the Clinical Global Assessment of Discharge Readiness—1 hour and 1.5 hours after dosing (the earliest discharge time point), and every 15 minutes thereafter until ready for discharge.
Local nasal tolerability was assessed by nasal examination and a self-report nasal symptom questionnaire (results are reported in the online supplement).
The Physician Withdrawal Checklist (
25) was administered to assess for potential withdrawal symptoms after cessation of esketamine treatment. Cognitive testing was performed to assess for potential impact on cognition; these data will be addressed in separate publications.
Statistical Analysis
Data were analyzed based on analysis sets that included all randomized patients who received at least one dose of intranasal study medication and one dose of oral antidepressant medication for the efficacy analyses, and at least one dose of either medication for the safety analyses.
Statistical tests were conducted at a two-sided significance level of 0.05. Analyses were performed using SAS, version 9.2.
Efficacy endpoints and analyses.
The primary efficacy endpoint, change in MADRS score from baseline (day 1) to endpoint (day 28), was analyzed using a mixed-effects model using repeated measures (MMRM). The model included baseline MADRS score as a covariate, and treatment, country, oral antidepressant class (SNRI or SSRI), day, and day-by-treatment interaction as fixed effects, and a random patient effect. A delta adjustment tipping point sensitivity analysis was performed to evaluate the robustness of the MMRM analysis to increasing deviations from the missing-at-random assumption (results are presented in the online supplement). Subgroup analyses were conducted according to the MMRM model used for the primary endpoint, including age, sex, baseline severity, antidepressant class, number of previous treatment failures, functional impairment, and region.
A serial gatekeeping (fixed sequence) approach was applied to adjust for multiplicity and to strongly control type I error across the primary and the three key secondary efficacy endpoints—namely, onset of clinical response by day 2, change in Sheehan Disability Scale score, and change in PHQ-9 score. These three key secondary endpoints were analyzed sequentially and considered significant at the two-sided 0.05 level only if the endpoint individually and previous endpoints in the hierarchy, including the primary endpoint, were significant at the two-sided 0.05 level.
The first key secondary efficacy endpoint compared the proportion of participants with onset of clinical response (defined as a ≥50% reduction in MADRS score by day 2 maintained to the end of the double-blind treatment phase with one excursion—i.e., a ≥25% reduction relative to baseline MADRS was allowed on day 8, 15, or 22) using a Cochran-Mantel-Haenszel chi-square test adjusting for country and antidepressant class.
The second and third key secondary efficacy endpoints—change from baseline to week 4 in Sheehan Disability Scale and PHQ-9 scores, respectively—were analyzed using the MMRM model described for the primary efficacy analysis but using the respective baseline score for the instrument as a covariate.
Other secondary efficacy endpoints included the proportions of responders (≥50% reduction from baseline MADRS score) and patients in remission (defined as a MADRS score ≤12) at the end of the 4-week double-blind treatment phase, change in CGI-S score, and anxiety symptoms (based on the Generalized Anxiety Disorder scale) and health-related quality of life and health status (based on the EQ-5D-5L).
In a post hoc analysis of response and remission rates, the number needed to treat (NNT) was estimated by taking the reciprocal of the risk difference. Change in Generalized Anxiety Disorder scale score was analyzed based on last-observation-carried-forward data using an analysis of covariance (ANCOVA) model, with country and antidepressant class as factors and baseline score as the covariate. The others were summarized descriptively.
Sample size determination.
The sample size planned for this study was calculated assuming a treatment difference for the double-blind treatment phase of 6.5 points in MADRS score between the esketamine plus antidepressant group and the antidepressant plus placebo group and a standard deviation of 12, based on the results of a phase 2 study of esketamine nasal spray for treatment-resistant depression (
11) and clinical judgment, a two-sided significance level of 0.05, and a dropout rate of 25%. Randomization of 98 individuals to each treatment group was required to achieve 90% power.
Results
Of 435 patients screened, nine (randomized at one site in Poland) were excluded from all analyses because of Good Clinical Practices violations (sensitivity analyses including this site confirmed no impact on the overall conclusions of the study), 227 underwent randomization to treatment; of these, three did not receive any study drug and one did not receive a dose of oral study antidepressant. Thus, the data set for the efficacy analyses included 223 patients (114 in the esketamine plus antidepressant group and 109 the antidepressant plus placebo group). Most randomized patients (197/227, 86.8%) completed the 28-day double-blind treatment phase (see Figure S1 in the online supplement).
The treatment groups were similar with respect to demographic and baseline clinical characteristics (
Table 1).
All patients in the esketamine plus antidepressant arm, with one exception (who received 42 mg because of a technical issue with the device), self-administered 56 mg esketamine on day 1 of the double-blind treatment phase. On day 4, just over half the patients in the esketamine plus antidepressant arm (58 of 107; 54.2%) remained at the 56 mg dose, and the remaining patients (49 of 107; 45.8%) were increased to the 84 mg dose. Two-thirds (66.7%) of the patients were on the 84 mg dose at the end of the 4-week treatment period. No patient requests to increase the esketamine dose or dosing frequency were reported in the study.
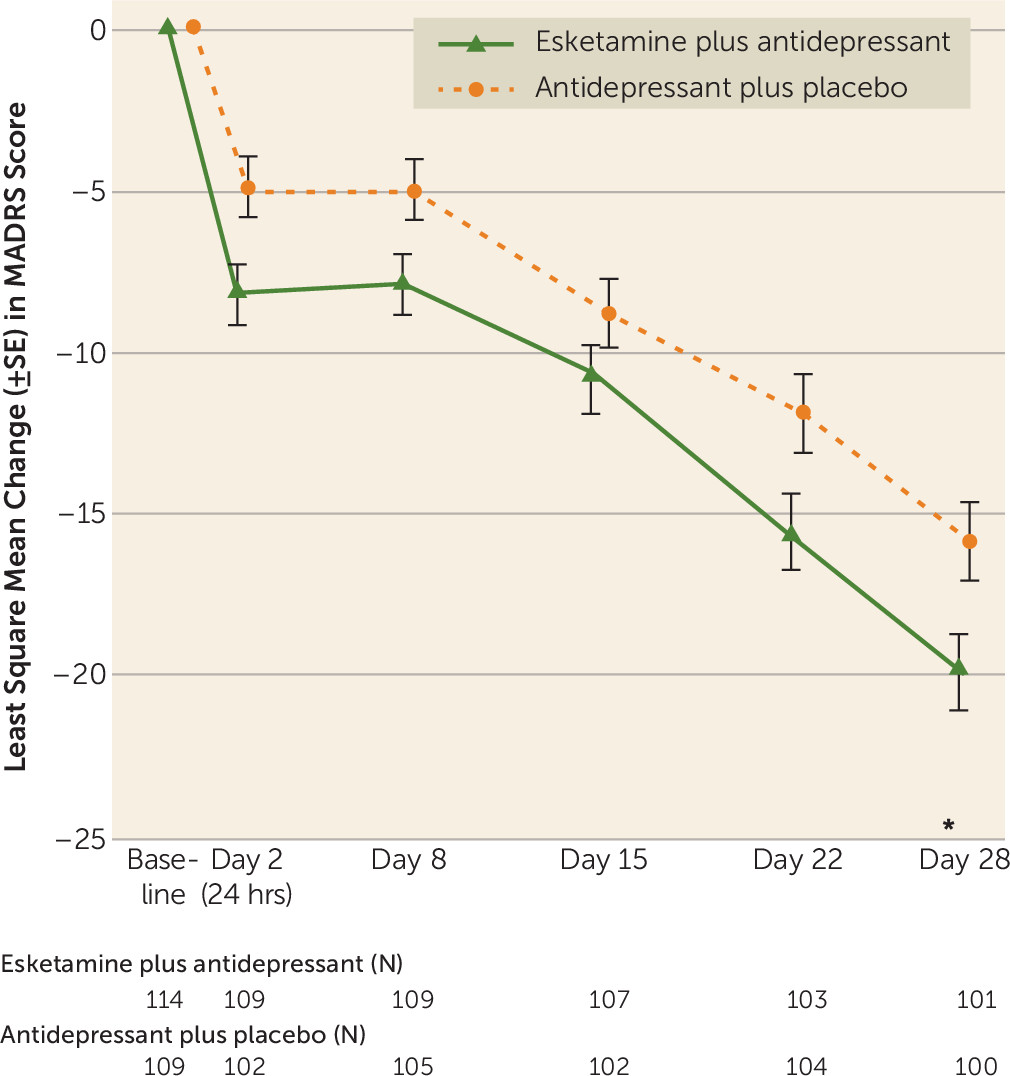
The mean MADRS score decreased from baseline to day 28, with greater improvement observed among those in the esketamine plus antidepressant arm as compared with the antidepressant plus placebo arm (difference of least square means=−4.0, SE=1.69, 95% CI=−7.31, −0.64; p=0.020) (
Table 2). The effect size for change in MADRS score from baseline to day 28 was 0.30. A scatterplot of individual patient data displaying MADRS score at baseline compared with day 28 is provided in Figure S2 in the
online supplement.
Response was rapid in onset and increased over time during repeated dosing, with least square mean between-group differences, favoring esketamine, of −3.3 (95% CI=−5.75, −0.85) 24 hours after dosing (i.e., at the day 2 visit), −2.9 (95% CI=−5.17, −0.59) at day 8, −2.0 (95% CI=−4.78, 0.82) at day 15, −3.8 (95% CI=−6.87, −0.65) at day 22, and, as noted above, −4.0 (95% CI=−7.31, −0.64) at day 28 (p=0.020) (
Figure 1).
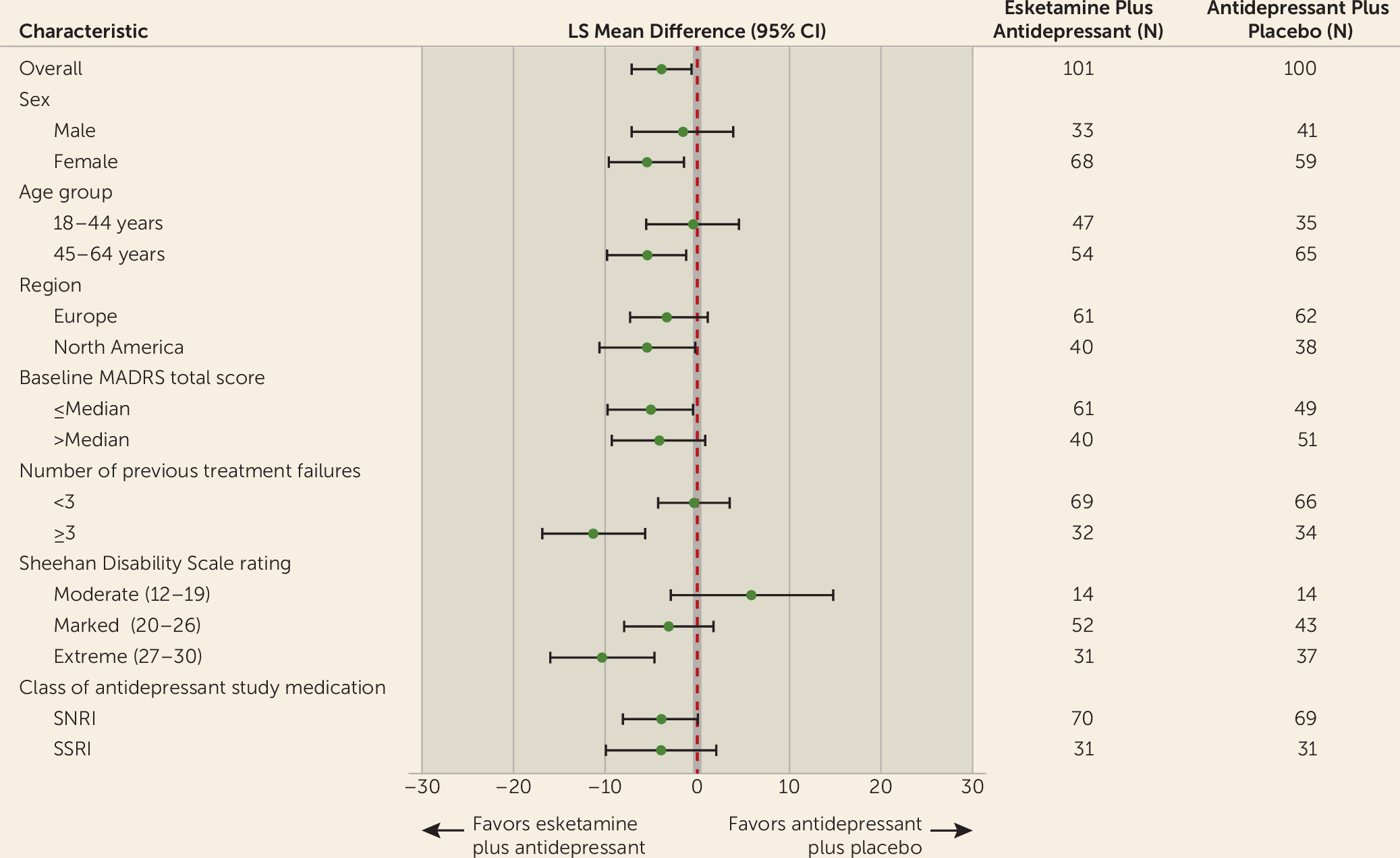
Subgroup analyses on the primary endpoint generally showed consistent benefit favoring esketamine in subgroups, including age, sex, region, baseline severity, number of previous treatment failures, functional impairment, and class of oral antidepressant (SNRIs and SSRIs) (
Figure 2).
In the hierarchical testing of the key secondary endpoints, the proportion of patients with a ≥50% decrease in MADRS score by day 2 (24 hours after a single dose) who also maintained this magnitude of reduction to day 28 (the first key secondary endpoint) was higher for the esketamine plus antidepressant arm compared with the antidepressant plus placebo arm (nine of 114 patients [7.9%] compared with five of 109 patients [4.6%]), although the difference was not significant (p=0.321) (see Table S5 in the online supplement). Early response (≥50% decrease in MADRS score by day 2) was observed in 18 of 109 patients in the esketamine plus antidepressant arm (16.5%), compared with 11 of 102 patients in the comparator arm (10.8%). Given the lack of statistical significance on the first endpoint, analyses of the other two key secondary endpoints could not be formally evaluated. Nevertheless, the results for change from baseline to day 28 in patient-reported Sheehan Disability Scale and PHQ-9 scores numerically favored the esketamine plus antidepressant arm over the comparator arm (difference of least square means of −4.0 [95% CI=−6.28, −1.64] and −2.4 [95% CI=−4.18, −0.69], respectively; see Table S5 in the online supplement).
In post hoc analyses, the proportion of patients who at a given time point were responders and in remission in both treatment groups generally increased over time during the double-blind treatment phase, with 70 of 101 patients (69.3%) in the esketamine plus antidepressant arm and 52 of 100 patients (52.0%) in the antidepressant plus placebo arm being responders at day 28 (odds ratio=2.4, 95% credible interval=1.30, 4.54). The NNT for response was 6. At day 28, 53 of 101 (52.5%) and 31 of 100 (31.0%) patients in the respective treatment groups were in remission, with an NNT of 5 for remission. No notable differences were observed between patients using concomitant benzodiazepines compared with those not using benzodiazepines.
Both treatment groups had a decrease in mean score on the Generalized Anxiety Disorder scale from baseline to the double-blind treatment phase endpoint (−7.9 [SD=6.12] from 13.2 at baseline in the esketamine plus antidepressant arm, and −6.8 [SD=5.75] from 13.1 at baseline in the antidepressant plus placebo arm (difference of least square means=−1.0, 95% CI=−2.35, 0.28).
Median CGI-S scores improved from baseline to endpoint of the double-blind treatment phase in both groups, with a median change from baseline of −2.0 (range=−5 to 1; interquartile range=2) in the esketamine plus antidepressant arm and −2.0 (range=−5 to 1; interquartile range=3) in the antidepressant plus placebo arm (see Table S6 in the online supplement). The ANCOVA analysis based on the ranks of change showed a numerical difference favoring the esketamine arm over the comparator arm, with an odds ratio of 2.8 (95% credible interval=1.14, 7.68), suggesting that the odds of an improved CGI-S score at endpoint for patients in the esketamine arm were 2.8 times the odds for patients in the comparator arm.
During the double-blind treatment phase, the five most common treatment-emergent adverse events observed among patients in the esketamine plus antidepressant arm were dizziness, dissociation, dysgeusia, vertigo, and nausea (incidences ranging from 20.9% to 26.1%); the incidence of each was two to 10 times lower in the antidepressant plus placebo arm (
Table 3). Most adverse events were of mild or moderate severity and were transient, with onset shortly after dosing and resolution by 1.5 hours after dosing, while patients were in the clinic. A total of 57.4% [66/115] and 10.1% [11/109] of patients in the esketamine and comparator arms, respectively, had a score ≤4 (moderate or greater sedation) on the Modified Observer’s Assessment of Alertness/Sedation at any postdose time during the double-blind treatment phase. Generally, for patients who experienced sedation, this symptom started around 15 minutes into dosing and peaked at 30 to 45 minutes after dosing, and symptoms of sedation spontaneously resolved by 1 to 1.5 hours after dosing. Sedation was not associated with hypoxemia.
One patient in the esketamine plus antidepressant arm experienced multiple injuries in a road traffic (motorbike) accident (reported as serious adverse events) on day 16 of the double-blind treatment phase (approximately 28 hours after an esketamine dose) and subsequently died on day 55, 40 days after the last dose of esketamine (additional information is provided in the online supplement). The investigator considered the events as doubtfully related to esketamine or to antidepressant. No other deaths or nonfatal study drug–related serious adverse events were reported.
Nine patients experienced one or more adverse events leading to discontinuation of intranasal study drug during the double-blind treatment phase, eight (of 115, 7.0%) in the esketamine plus antidepressant arm (single events of anxiety, depression, depressive symptoms, panic attack, drug intolerance, feeling drunk, dizziness, headache, vertigo, nausea, road traffic accident, and multiple injuries) and one (of 109, 0.9%) in the antidepressant plus placebo arm (generalized rash).
Transient blood pressure increases occurred after each dose of esketamine; the maximum value was reached at 40 minutes after dosing in most cases and typically returned to or near the predose range by 1.5–2 hours after dosing (see Figure S3 in the online supplement). The maximum across dosing days of the mean maximum increase in systolic blood pressure from before dosing at any postdose time point was +11.6 mmHg in the esketamine plus antidepressant arm and +5.0 mmHg in the antidepressant plus placebo arm, and in diastolic blood pressure was +8.1 and +4.5 mmHg for the respective treatment groups.
No clinically significant change in ECG was observed during the study.
The percentage of patients reporting suicidal ideation (C-SSRS scores of 1 [wish to be dead], 2 [nonspecific active suicidal thoughts], or 3 [active suicidal ideation with any methods, not plan, without intent to act]) decreased from baseline to the endpoint in both treatment groups (see Figure S4 in the online supplement). In both treatment groups, the maximum score reported on the C-SSRS during the double-blind treatment phase was 3. Six of 112 (5.4%) patients in the esketamine plus antidepressant arm and seven of 109 (6.4%) patients in antidepressant plus placebo arm had treatment-emergent postbaseline suicidal ideation. No patients in either treatment group had treatment-emergent suicidal behavior.
Present-state dissociative symptoms and transient perceptual effects measured by the CADSS (see Figure S5 in the online supplement) began shortly after the start of esketamine dosing, peaked at 40 minutes, generally resolved by 1.5 hours, and attenuated with repeated dosing (the mean change in CADSS score from before dosing to 40 minutes after dosing decreased from 7.8 [SE=0.84] on day 1 to 3.5 [SE=0.55] on day 25). No symptoms or adverse events of psychosis were reported. The number needed to harm for dissociation was 5 for the esketamine plus antidepressant arm compared with the antidepressant plus placebo arm. The proportion of responders at day 28 was comparable for patients who experienced dissociation (maximum CADSS score >4 on day 1) and those who did not (maximum CADSS score ≤4 on day 1), with response rates of 68.5% and 70.2%, respectively.
On each intranasal treatment day, approximately half (≥44.3%) of the patients in the esketamine plus antidepressant arm and ≥92.0% of those in antidepressant plus placebo arm were considered ready for discharge (based on overall clinical status, including sedation, blood pressure, and other adverse events) by 1 hour after dosing, based on the Clinical Global Assessment of Discharge Readiness, and more than 90% in each treatment group by 1.5 hours after dosing (93.2% and 98.9% for the esketamine and comparator arms, respectively). All patients were ready for discharge by 3 hours.
Changes in withdrawal symptoms assessed by the Physician Withdrawal Checklist after cessation of esketamine were consistent with observed changes in symptoms of depression and anxiety. The most frequently reported (≥25%) new or worsened symptoms among patients in the esketamine plus antidepressant arm were fatigue-lethargy/lack of energy, weakness, dysphoric mood/depression, loss of appetite, and restlessness/agitation, and for patients in the antidepressant plus placebo arm, insomnia, irritability, and dysphoric mood/depression. No clear evidence of withdrawal was observed at either 1 or 2 weeks after cessation of treatment with esketamine nasal spray. There were no reports of drug abuse or cravings during the follow-up phase.
Discussion
In this study, patients with treatment-resistant depression achieved clinically meaningful and statistically significant improvement (based on change in MADRS score after 28 days) in depressive symptoms after being switched to esketamine nasal spray plus a newly initiated oral antidepressant, with a group treatment difference of −4.0 against an active comparator—a newly initiated antidepressant plus intranasal placebo.
While the hypothesized between-group difference of −6.5 was not reached, this large treatment difference had been based on previously conducted phase 2 esketamine treatment-resistant depression studies (
10,
11) and not on any threshold for defining a clinically meaningful improvement; the observed −4.0 difference exceeded minimum clinically important difference thresholds reported in the literature (
26,
27). The between-group difference in this study is similar to that on the same primary outcome observed in two other phase 3 short-term studies of esketamine for treatment-resistant depression—a fixed-dose study in the same age group (56 mg: −4.1; 84 mg: −3.2] (
28) and a study of patients ≥65 years old (−3.6) (
29) conducted at the same time, although statistical significance was not achieved in those two studies. Of note, the mean MADRS treatment differences for adjunctive brexipiprazole (
30), quetiapine (
31), or aripiprazole (
32) in major depression and for combination treatment with olanzapine/fluoxetine (
33) in treatment-resistant depression range from −1.94 to −3.17.
The number of patients meeting the full criterion for onset of clinical response by day 2 with persistence to day 28, a metric not evaluated in previous antidepressant drug or ketamine trials, was lower than anticipated. Factors potentially contributing to this finding were 1) the strict criteria used, which included onset and sustained response through the entire 4-week treatment period and excluded patients who had any fluctuation in scores early in the treatment, a finding commonly seen over the course of treatment in depression; and 2) the use of independent, remote MADRS raters, which may have reduced the sensitivity of detecting the onset of clinical response by day 2, as remote raters did not know individual patients and their baseline characteristics, could only assess reported symptoms on the structured interview, and could not take into account any of the signs of improvement of depression.
Earlier studies of esketamine in patients with treatment-resistant depression (
10,
11) and major depression at imminent risk for suicide (
12), which demonstrated greater magnitude of early response, applied a less stringent definition for response onset and employed site-based raters for efficacy evaluation.
Although the endpoint for onset of clinical response was not statistically significant, the difference in least square mean MADRS score between treatment groups at day 2 (24 hours after a single dose) was −3.3 points, which is considered a clinically meaningful treatment difference (
26,
27) suggesting rapid onset of antidepressant efficacy. Moreover, the mean difference between the active and control arms observed at day 2 generally persisted through day 28 (
Figure 1).
Based on the predefined testing sequence, the other two key secondary endpoints (change in Sheehan Disability Scale and PHQ-9 scores) could not be formally evaluated. However, in post hoc analyses, the results from both patient-reported outcomes showed consistency with the primary clinician-based outcome result (see Figure S6 in the online supplement), providing supportive evidence from patients’ perspective of improvement in mood and function after 4 weeks of treatment.
The response and remission rates seen in the antidepressant plus placebo arm were higher than those reported in the Sequenced Treatment Alternatives to Relieve Depression trial for step 3 (response rate, 16.8%; remission rate, 13.7%) and in the olanzapine-fluoxetine study (
33) (response and remission rates, respectively, of 40.4% and 27.3% for olanzapine/fluoxetine combination, 29.6% and 16.7% for fluoxetine, and 25.9% and 14.7% for olanzapine). A number of factors may have contributed to the smaller than anticipated effect size finding, the lower treatment difference compared with our phase 2 work, and the higher response and remission rates observed for the oral antidepressant plus placebo group: 1) use of a newly initiated antidepressant (to which the patients had not shown a previous nonresponse) in the comparator arm (i.e., not a true placebo control), a design element that would increase expectation of response in the active comparator arm; 2) high frequency of patient interaction with clinic staff because of the need for twice-weekly visits; 3) use of a nasal spray delivery system, leading to patients’ expectation of “something novel”; 4) high patient expectation of benefit as a result of the portrayal in the media of ketamine as a new treatment option for depression; and 5) nocebo response (i.e., adverse effect following an “inert” treatment), as noted by an increase in CADSS scores after placebo nasal spray administration to which a bittering agent was added for blinding.
The odds ratio of 2.4 for response at day 28 in the esketamine plus oral antidepressant group was higher compared with odds ratios reported in a recent systematic review of antidepressant efficacy (
34). The NNT for the esketamine plus oral antidepressant arm was 6 for response and 5 for remission. The only medication approved for the treatment of treatment-resistant depression is olanzapine plus fluoxetine, which has an NNT of 8 for response and 13 for remission (
35). NNT for response and remission provide clinical perspective for an effect size of 0.3 on the primary endpoint.
Analyses of dissociation/perceptual change symptoms (assessed by the CADSS) and sedation (assessed by the Modified Observer’s Assessment of Alertness/Sedation) suggest onset shortly after esketamine administration, with resolution generally by 1.5 hours after dosing, and evidence of attenuation of dissociation after repeated administrations, unlike esketamine’s antidepressant efficacy, which was sustained. The magnitude of postdose blood pressure increases was lower compared with the previous phase 2 study of esketamine nasal spray (
11), possibly because of the risk-mitigation blood pressure guidelines initiated in phase 3 studies (see the
online supplement). Most patients were ready for discharge by 1.5 hours after dosing, with the latest discharge readiness time point reported at 3 hours.
Because this trial was a flexible-dose study, dose-response relationships were not evaluated. The trial aimed to assess the short-term efficacy and safety of esketamine, and therefore it does not inform on maintenance of effect and long-term safety, which are being evaluated in other studies (
36,
37).
In this trial, esketamine efficacy was evaluated when combined with a newly initiated antidepressant. To further inform clinical practice, trials of monotherapy or of esketamine augmentation to an ongoing antidepressant may be considered in the future.
Some limitations of the study design merit comment. The generalizability of the study findings may be limited by the exclusion of patients with significant psychiatric or medical comorbidities or substance dependence, non-treatment-resistant forms of major depression, prior nonresponse to ketamine or esketamine in the current episode, and imminent risk of suicide (studied in a separate program) as well as a low proportion of nonwhite patients. While eventual efficacy bias was mitigated by using independent remote MADRS raters, it is possible that the specific adverse event profile of esketamine affected the blind for the study patients. Although patients were not specifically asked whether they believed they had received drug or placebo, it is noteworthy that the dissociation ratings (CADSS scores) increased in the control group who received placebo nasal spray, providing evidence of adequate blinding and supporting expectation of benefit as one of the contributing factors of the higher than anticipated placebo response observed in the control group.
In summary, despite the unexpected high response to the oral antidepressant plus placebo comparator, results of the study consistently showed a clinically relevant, favorable improvement in depressive symptoms with esketamine (either 56 mg or 84 mg) nasal spray plus a newly initiated antidepressant as assessed by change in MADRS score after 28 days in adult patients with treatment-resistant depression and clinically meaningful benefit 24 hours after the first dose. Moreover, administration of esketamine in this sample appeared safe and tolerated.