The majority of the first 12 months of life is spent asleep (
1). Critical brain maturation processes are thought to occur during sleep in early development (
2,
3)—for example, development of the visual cortex relies on both sensory stimulation during wakefulness and endogenous stimulation during sleep, which together guide neuronal differentiation and developmentally regulated synaptic plasticity (
4). Children with autism spectrum disorder (ASD) are 2–3 times more likely to have difficulties with initiating or sustaining sleep than typically developing children (
5). The impact of inadequate sleep on child cognitive development, behavior, and family functioning in ASD is evident in the behavioral literature (
6–
8). However, to our knowledge, no research has examined the effect of poor sleep on brain development in this population.
Reduced sleep duration in children with ASD compared with typically developing children has been reported as early as 30 months of age (
9) and can persist through adulthood (
7). Sleep patterns in ASD may diverge even earlier. A recent study of more than 1,000 children found that the number of night awakenings at 12 months of age was associated with ASD screening scores at age 24 months (
10). Emerging evidence suggests that preclinical brain and behavior differences exist during infancy in children later diagnosed with ASD. Atypical sensorimotor development (
11) and accelerated brain growth in the first year of life have been shown to precede the consolidation of ASD symptoms (
12,
13). Given that a substantial portion of an infant’s time is spent asleep, it is possible that some of the preclinical differences seen in early neurodevelopment in ASD could be related to atypical sleep.
Our aim in this study was to characterize altered sleep patterns and associations with brain development in a sample of infants at high familial risk (i.e., having an older sibling with ASD) or low familial risk for ASD (i.e., no family history of ASD in first-degree relatives). This sample provided an opportunity to characterize sleep difficulties in infants who later develop ASD and between those at high and low risk. We examined the relationship between sleep during infancy and subcortical volume during early development. We chose to focus on subcortical development because altered volume in these structures (i.e., the hippocampus, amygdala, thalamus, and basal ganglia) has been associated with ASD in numerous studies (reviewed in
13–
15) and also with sleep problems. Measures of insomnia severity in adults (i.e., arousal level, sleep latency, quality, and fragmentation) have been associated with decreased hippocampal (
16–
18) and putamen volumes (
18). The literature on brain correlates of pediatric sleep problems is limited. Sleep duration during weekdays in typically developing children has been found to be positively associated with bilateral hippocampal volume, after adjusting for age, sex, and intracranial volume (
19). A recent prospective neuroimaging study found that trajectories of sleep disturbance from age 2 months to 6 years were associated with smaller total brain volumes by age 7; however, subcortical volumes were not significantly different after correcting for total volume (
20). Therefore, whether sleep disturbance in early childhood is associated with morphometric changes in subcortical structures remains an open question—one well suited to investigation in the context of ASD.
We examined associations between sleep difficulties and developmental trajectories in six subcortical structures that are altered in ASD: the amygdala, hippocampus, caudate, putamen, globus pallidus, and thalamus (
13–
15). Based on available evidence of early sleep differences associated with ASD screening scores (
10), we hypothesized that sleep difficulties in the first 12 months of life would occur more often among high-risk infants who go on to develop ASD (high-risk ASD), least often among low-risk infants, and at an intermediate level among high-risk infants who do not develop ASD (high-risk non-ASD) (
21). We further hypothesized that poor sleep would be related to alterations in subcortical brain morphometry, a hypothesis that was nondirectional, given the paucity of data regarding the impact of sleep difficulties on brain morphometry in early development. Additional follow-up analyses were conducted to determine whether hemispheric differences, cognitive ability, infant temperament, and the timing of sleep measurement were significant contributors to the sleep-brain relationships we observed.
Results
ISOP Scores Across Diagnostic Groups
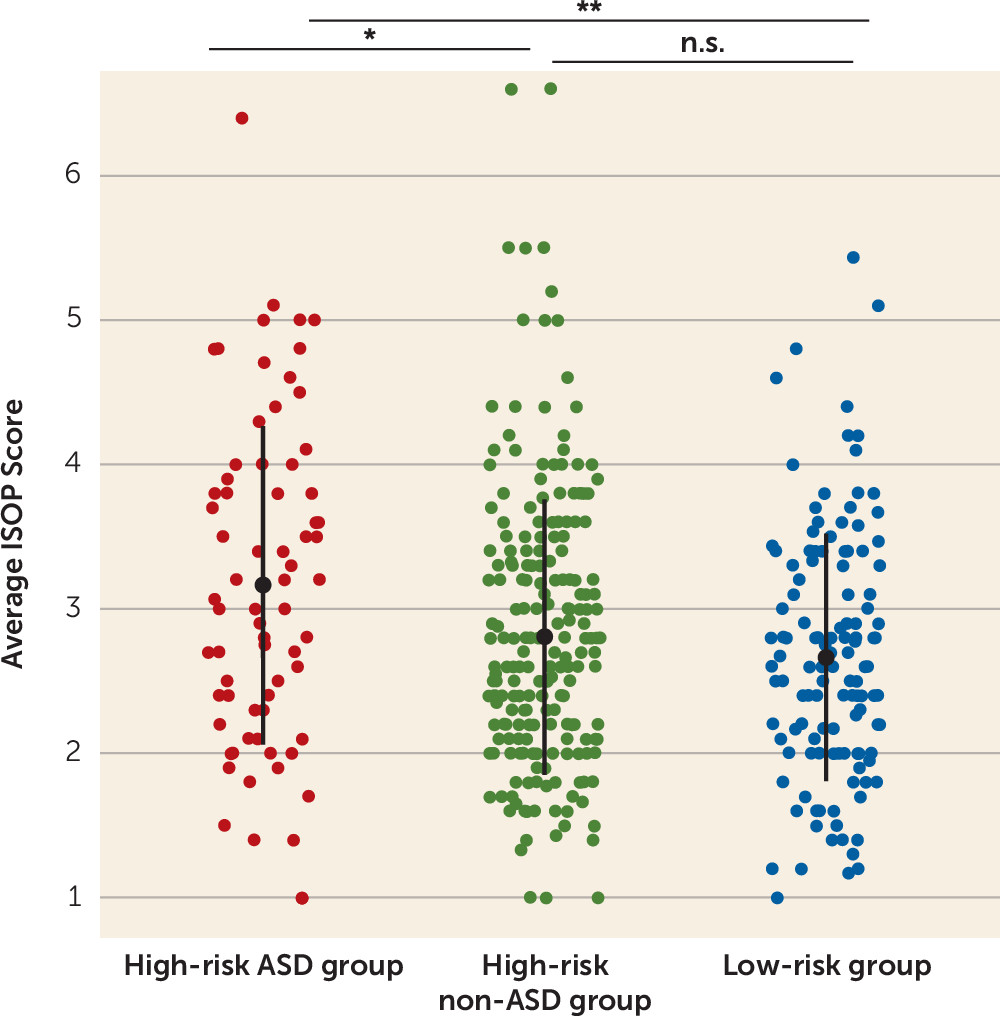
ISOP scores were compared across the three diagnostic groups and differed significantly (F=6.41, df=2, 429, p=0.002) (
Figure 2). A post hoc Tukey test revealed greater sleep onset problems in the high-risk ASD group compared with the high-risk non-ASD group (difference, 0.36; 95% CI=0.06, 0.66, p=0.02) and the low-risk group (difference, −0.50; 95% CI=−0.83, −0.17, p=0.001). ISOP scores did not differ significantly between the high-risk non-ASD and low-risk groups (difference, −0.14; 95% CI=−0.39, 0.10, p=0.36). A disorder-specific effect was also found when examining ISOP scores from the 6-month time point only (high-risk ASD group compared with high-risk non-ASD group: t=2.72, df=82.7, p=0.008; high-risk ASD group compared with low-risk group: t=3.0, df=89.0, p=0.003; high-risk non-ASD group compared with low-risk group: t=0.53, df=272.3, p=0.60) and the 12-month time point only (high-risk ASD group compared with high-risk non-ASD group; t=2.00, df=90.9, p=0.04; high-risk ASD group compared with low-risk group: t=3.0, df=108.4, p=0.004; high-risk non-ASD group compared with low-risk group: t=1.62, df=229.4, p=0.11). It is noteworthy that there was no difference in ISOP scores between male and female infants in the study sample (F=0.10, df=1, 430, p=0.75).
Given the finding of categorical differences in ISOP scores, we explored associations between ISOP score and continuous measures of cognitive and adaptive development and ASD symptoms. Correlation results are presented in Table S2 in the online supplement. At each time point (6 months and 12 months), ISOP score correlated significantly with expressive language scores on the Mullen Scales of Early Learning (6 months, r=−0.14, p=0.01; 12 months, r=−0.11, p=0.03) but not with any of the other behavioral measures. By 24 months, average ISOP scores were significantly correlated with Autism Diagnostic Observation Schedule social affect severity scores (r=0.14, p=0.004), Mullen expressive language scores (r=−0.11, p=0.03), Vineland Adaptive Behavior Scale communication scores (r=−0.13, p=0.01), and Vineland socialization scores (r=–0.15, p=0.003). Across the entire sample, infants with worse sleep from 6 to 12 months showed weaker social communication skills by 24 months.
Sleep Onset Problems in Relation to Brain Volume Trajectories From 6 to 24 Months
Separate linear mixed-effects models were conducted for each subcortical structure (see Table S3 in the
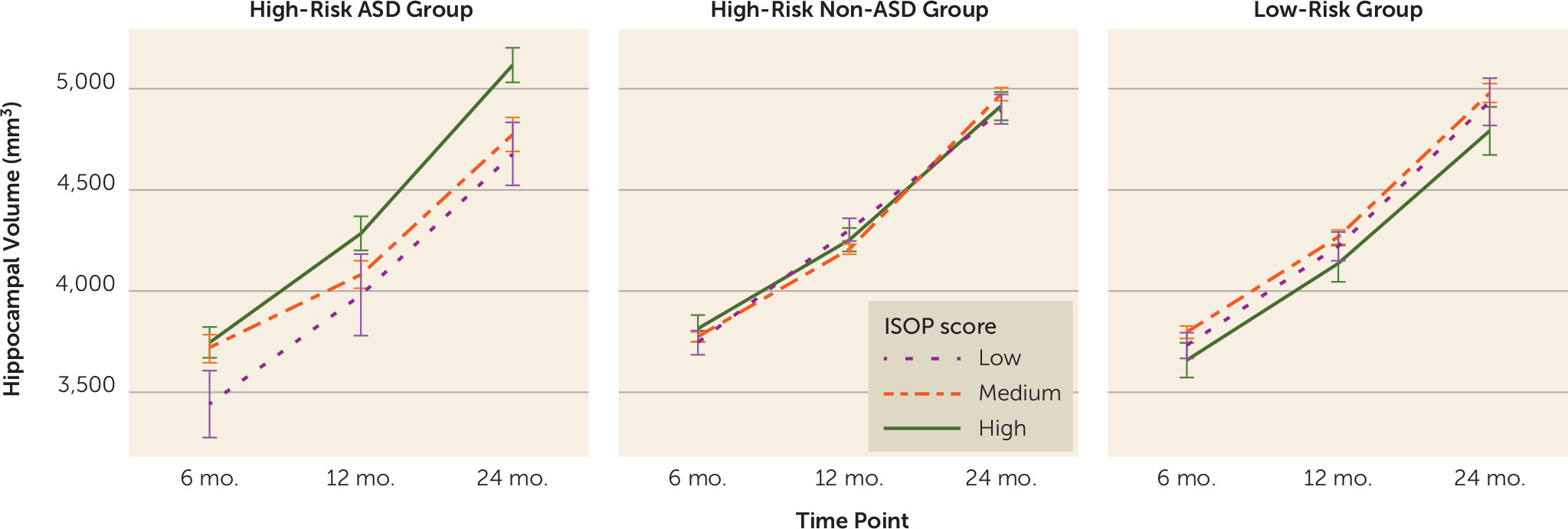
online supplement). Only hippocampal trajectory was significantly predicted by ISOP score (β=101.0, t=3.2, p=0.008), and group moderated the association between ISOP score and hippocampal trajectory in infancy (
Figure 3). Examination of group-specific parameter estimates revealed that sleep onset difficulties in infancy were associated with increased hippocampal volume from 6 to 24 months only for high-risk siblings who went on to develop ASD (β=104.7; t=2.8, p=0.006); no relationship between sleep and hippocampal trajectory was found in the high-risk non-ASD group (β=−11.5; t=0.6, p=0.6) and the low-risk group (β=−4.9; t=0.2, p=0.9).
Hemispheric Differences
Examining associations in the left and right hemispheres separately revealed that ISOP score was significantly associated with trajectories of both left and right hippocampal volume. Specifically, there was a significant main effect of ISOP score (left: β=47.9, t=3.0, p=0.002; right: β=53.1, t=3.1, p=0.002) and significant group-by-ISOP interactions for each hemisphere (left: high-risk ASD group compared with high-risk non-ASD group: β=−49.9, t=2.7, p=0.007; high-risk ASD group compared with low-risk group: β=−54.5, t=2.5, p=0.012; right: high-risk ASD group compared with high-risk non-ASD group: β=−59.0, t=2.9, p=0.003; high-risk ASD group compared with low-risk group: β=−54.3, t=2.3, p=0.02).
Hippocampal Trajectory and Sleep Onset Problems, Controlling for Cognitive Ability
The association between ISOP score and hippocampal trajectory persisted after adding 24-month cognitive ability (Mullen early learning composite) scores to the model. Specifically, there was a significant main effect of ISOP score (β=100.0, t=3.1, p=0.002) and a significant group-by-ISOP interaction (high-risk ASD group compared with high-risk non-ASD group: β=−106.7, t=2.9, p=0.005; high-risk ASD group compared with low-risk group: β=−111.7, t=2.5, p=0.012). Twenty-four-month scores on the Mullen scales did not predict additional variance in hippocampal trajectory (β=−0.8, t=0.9, p=0.37).
Hippocampal Trajectory and Other IBQ-R Subscales
The IBQ-R has 14 subscales (see reference
22). The five sleep items that comprise the ISOP are part of the falling reactivity/rate of recovery from distress scale. To examine the specificity of the sleep-hippocampal finding, the same full linear mixed model predicting bilateral hippocampal trajectory was repeated, replacing the ISOP score with scores from each IBQ-R subscale (including the full 13-item falling reactivity/rate of recovery from distress scale, each averaged from 6 to 12 months). None of the other IBQ-R subscales were significantly related to hippocampal volume (see Table S4 in the
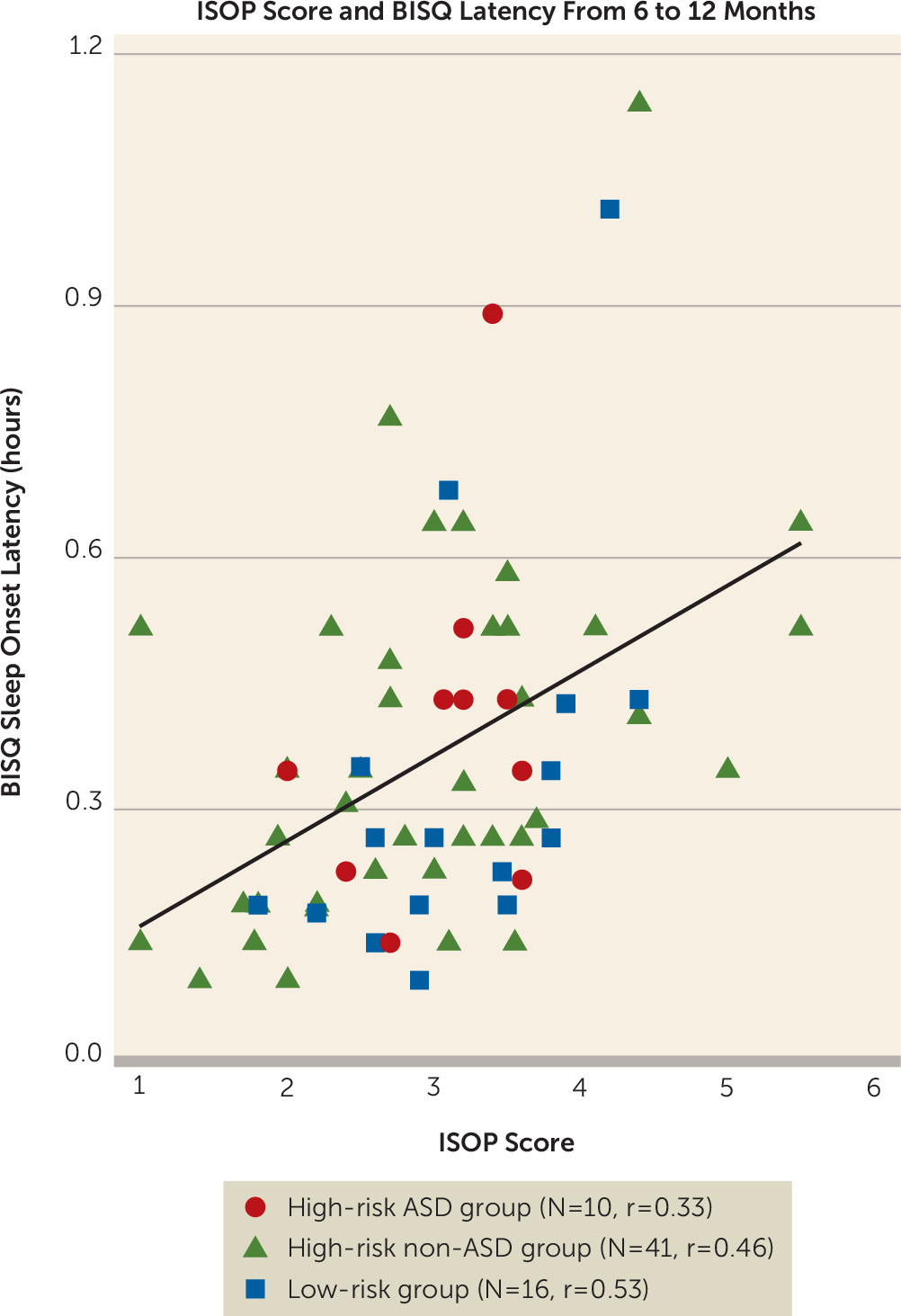
online supplement), including the full falling reactivity/rate of recovery from distress scale from which the sleep items were derived. This suggests that the relationship with hippocampal trajectory may be specific to the five IBQ-R items comprising the ISOP score and does not represent a more general relationship with infant behavior or temperament.
Hippocampal Trajectory and Sleep Onset Problems Measured at 6 and 12 Months
The final follow-up analyses investigated whether the associations between ISOP score and trajectories of hippocampal volume from 6 to 24 months would persist if restricted to sleep measured at a single time point in development (either 6 or 12 months). Models were run substituting 6-month and 12-month ISOP scores for the averaged score and including cognitive ability (Mullen early learning composite score) measured at that same time point (6 or 12 months).
The association between hippocampal volume trajectory and sleep persisted when we used ISOP scores measured only at a single time point (6 or 12 months). Specifically, in the 6-month model, there was a significant main effect of 6-month ISOP score (β=75.9, t=2.3, p=0.02) and a significant group-by-6-month ISOP score interaction (high-risk ASD group compared with high-risk non-ASD group: β=−78.4, t=−2.1, p=0.04; high-risk ASD group compared with low-risk group: β=−109.8, t=2.6, p=0.008). Six-month scores on the Mullen scales did not predict additional variance in hippocampal trajectory (β=0.8, t=0.6, p=0.6).
Similarly, in the 12-month model, there was a significant main effect of 12-month ISOP score (β=74.5, t=2.4, p=0.02) and a significant group-by-12-month ISOP score interaction for the contrast of the high-risk ASD group compared with the high-risk non-ASD group (β=−86.9, t=−2.4, p=0.02). With the 12-month ISOP score, the contrast of the high-risk ASD group compared with the low-risk group did not reach statistical significance (β=−43.9, t=−1.1, p=0.3). Twelve-month scores on the Mullen scales did not predict additional variance in hippocampal trajectory (β=−0.22, t=0.20, p=0.8).
Discussion
In a large sample of infants at high and low familial risk for ASD, we found that sleep onset problems were more prevalent at 6–12 months among infants who went on to develop ASD. Difficulties with sleep onset during infancy were associated with weaker social communication skills by 24 months and were related to hippocampal volume trajectory from 6 months to 24 months only for the high-risk ASD group. Infants in the high-risk ASD group who had sleep onset difficulties at 6–12 months exhibited increased hippocampal volume trajectories compared with those in the high-risk ASD group who had better sleep. The association with sleep onset problems in infancy was specific to the hippocampus; no significant sleep-brain associations were found for the other subcortical structures examined (the amygdala, caudate, globus pallidus, putamen, and thalamus). Additional analyses revealed that the observed association between sleep onset problems and hippocampal trajectories in the high-risk ASD group existed in each hemisphere and persisted after controlling for cognitive ability at 24 months. In addition, the association was specific to sleep onset problems and did not generalize to other aspects of infant temperament measured on the IBQ-R. Finally, sleep-hippocampal trajectory associations persisted in models that included sleep and cognitive ability measured at a single time point (6 or 12 months).
Our finding that early sleep onset problems are specific to ASD is in line with findings from previous infant sibling studies. Sleep patterns in typically developing children who have a sibling with ASD are similar to those with no family history of ASD (
21,
25). Associations with hippocampal trajectories were also specific to the high-risk ASD group. In contrast to previous studies with typically developing older children, with adults with insomnia, and with sleep-deprived animals (
18,
19,
26), sleep onset problems in infants at high risk who developed ASD were associated with increased (rather than decreased) hippocampal volume across early development. The reason for this disorder-specific effect and its directionality is unclear, but it may be related to a disrupted coupling of hippocampal and brain sizes that has been observed in other ASD samples (
27). Overgrowth of some structures, but not others, at different stages of development has been frequently reported in ASD (
12,
13,
28). Indeed, in the same infant sibling sample as the present study, early cortical overgrowth was associated with later social deficits (
12), larger corpus callosum volumes were associated with later restricted or repetitive behavior (
29), and larger thalamus, caudate, and amygdala volumes were associated with abnormal language profiles (
24). In a departure from this previous work, however, we show evidence of a brain-behavior relationship during infancy that precedes the onset of ASD symptoms. If replicated, these findings suggest that an individual difference in infant behavior (i.e., sleep onset difficulties) could help predict which infants will show abnormal trajectories of hippocampal growth before the onset of ASD symptoms.
The demonstration that our findings are specific to the hippocampus is consistent with a large body of non-ASD research linking sleep to morphometric changes (
16–
18) and accumulation of metabolic by-products in the hippocampus (e.g., beta-amyloid [
30]). Sleep also plays a role in hippocampus-mediated cognitive processes, including spatial and declarative learning and consolidation (
31). Adequate sleep may be required for typical hippocampal maturation: young (but not adolescent) mice that were deprived of REM sleep showed reduced long-term potentiation stability and lower expression of glutamatergic signaling proteins, both suggestive of altered hippocampal development (
32). Thus, the hippocampus appears to be a brain region that is particularly sensitive to the effects of disturbed or inadequate sleep. However, it is important to recognize that the sleep-hippocampus associations we observed do not reveal a causal direction. Inadequate sleep may cause changes in the hippocampus that confer vulnerability to disorders of neurodevelopment or neurodegeneration. Alternatively, preexisting neurobiological or genetic differences may result in both hippocampal changes and sleep disturbance during the early stages of a disorder.
A variety of critical neurobiological processes occur during sleep and could underlie our findings. One potential mechanism involves neuroinflammatory processes that are modulated during sleep. Sleep deprivation has been shown to increase neuroinflammation, which affects synaptic plasticity in the hippocampus (
33). Sleep disturbance has been associated with inflammatory responses in the hippocampus, but not the cortex, in adult mice and impaired subsequent performance on a hippocampus-dependent learning and memory task (
34). In humans, the data on associations between sleep and inflammation are mixed. A recent meta-analysis found that increased levels of inflammatory markers were associated with sleep disturbance and long sleep duration (>8 hours per night) but not short sleep duration (<7 hours per night [
35]). However, evidence of potential involvement of inflammatory responses in the pathogenesis of ASD (
36) suggests that investigating sleep and neuroinflammation longitudinally in studies of early brain development (such as the present study) is a promising direction for future research.
This study has several limitations, including the use of a novel measure of sleep during infancy. Although our measure of sleep onset problems (derived from five items on the IBQ-R) revealed strong correspondence with BISQ scores and strong internal consistency in a subsample of infants, it was based on parent report and has not (to our knowledge) been validated against objective measures of sleep in infancy (i.e., actigraphy or polysomnography). In addition, these five items measured only one aspect of sleep (sleep onset latency) and did not provide information about sleep fragmentation, quality, or overall duration. It will be important in future studies to conduct more comprehensive sleep assessments to determine which characteristics of sleep during infancy are most relevant to brain and behavior development. Another important area for future research will be to investigate associations between infant sleep and cortical development, because altered cortical morphometry has been associated with ASD in high-risk siblings (
12) and chronic sleep disturbance in typically developing children (
20). Given the early stages of this study, and the absence of previous work examining sleep and neurodevelopment in infants at high risk for ASD, we believe these preliminary findings are of interest and warrant extension to other brain regions and replication with alternative measures of sleep.
If replicated, these observations could provide new insights into a potential role for disturbed sleep in the development of ASD. The high prevalence of ASD and comorbid sleep difficulties in some rare genetic syndromes (37) has led to the suggestion that ASD and sleep problems may have shared etiology (38). Impairments in circadian timing (39), sleep-wake regulation (8), sensory processing (38), and affective functioning (7) may underlie both sleep difficulties and core symptoms of ASD. The efficacy of both pharmacologic (e.g., melatonin) and behavioral sleep interventions for some children with ASD suggests that both neurobiological and psychosocial factors should be considered. Our findings provide initial evidence that sleep difficulties in the first year of life may precede ASD diagnosis and are associated with altered neurodevelopmental trajectories in high-risk siblings who go on to develop ASD. We expect future work to reveal the implications of these results for understanding neurodevelopment in ASD and for developing early, targeted interventions for sleep difficulties in infants at high risk for ASD.