We hypothesized that both high baseline SVD severity and a higher rate of SVD progression are independently associated with an increased risk of all-cause dementia, especially for the incidence of vascular dementia.
Methods
Study Design and Population
This study is embedded within the long-term Radboud University Nijmegen Diffusion Tensor and Magnetic Resonance Cohort (RUN DMC) study in individuals with sporadic SVD, which investigates the risk factors and clinical outcomes of SVD. The study rationale and protocol have been reported previously (
11). In short, a total of 503 consecutive community-dwelling patients, ages 50–85 years, who were referred to the Radboud University Medical Center neurology outpatient clinic and who underwent brain MRI were screened for baseline inclusion in 2006. SVD diagnosis was based on MRI and included presence of WMHs or lacunes (
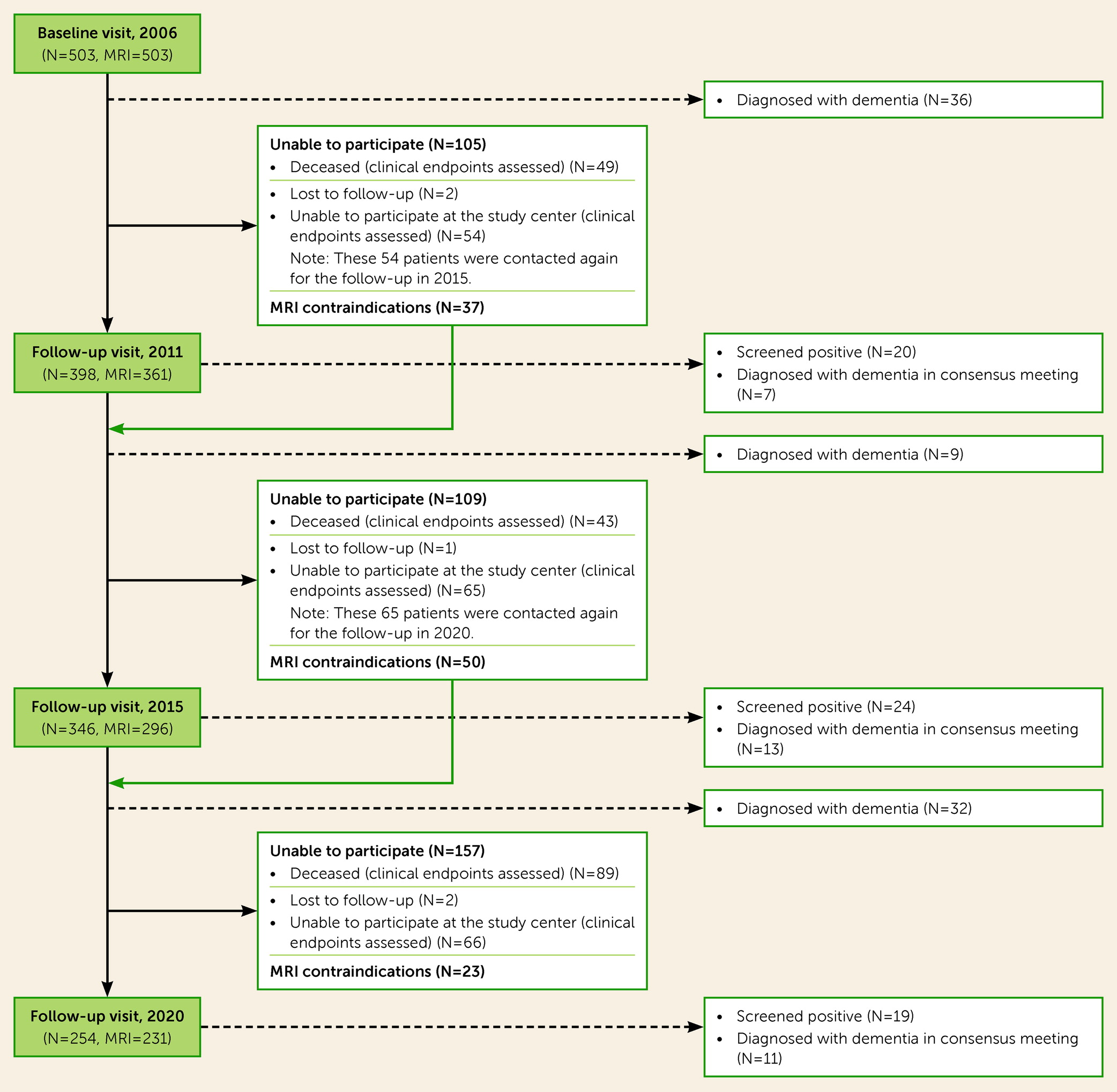
12). The main exclusion criteria at baseline were presence of dementia, parkinsonism, non-SVD-related white matter lesions (e.g., multiple sclerosis), or a life expectancy of less than 6 months. Follow-up assessments took place in 2011, 2015, and 2020. At all time points, patients completed structured medical questionnaires and underwent physical examination, motor tests, standardized neuropsychological examination, and brain MRI. A flowchart of patient participation in all follow-ups is provided in Figure S1 in the
online supplement.
The Medical Review Ethics Committee region Arnhem‐Nijmegen approved the study, and all participants gave written informed consent.
Neuropsychological Examination and Education Classification
Cognitive function at baseline and at all follow-up waves was assessed by a standardized neuropsychological test battery covering the following cognitive domains: working and episodic memory, executive function, psychomotor speed, fluency, and attention. The cognitive tests have been described in detail elsewhere; parallel versions were used when appropriate to overcome material-specific practice effects (
13).
Education level was classified into seven levels in accordance with the Dutch educational system (1 indicates having less than a complete primary education and 7 indicates having a university degree or higher). We then dichotomized these into two categories, namely, primary education or less (levels 1–2) and more than primary education (levels 3–7).
Vascular Risk Factors
Hypertension was defined as a systolic blood pressure ≥140 mmHg and/or a diastolic blood pressure ≥90 mmHg at the baseline examination or the use of blood-pressure-lowering medications. Diabetes and hypercholesterolemia were considered to be present if the patient was taking oral glucose-lowering drugs, insulin, or lipid-lowering drugs. Information on smoking status was dichotomized into ever (current or former) or never smoking.
MRI Protocol
Images were acquired on 1.5-T MRI scanners (in 2006, Magnetom Sonata [Siemens]; in 2011 and 2015, Magnetom Avanto [Siemens]) and included the following whole-brain scans: three-dimensional T
1 MPRAGE imaging (isotropic voxel size, 1.0 mm
3); FLAIR sequences (2006: voxel size=0.5×0.5×5.0 mm, interslice gap=1.0 mm; 2011 and 2015: voxel size=0.5×0.5×2.5 mm, interslice gap=0.5 mm); and a transversal T
2*-weighted gradient echo sequence (2006: isotropic voxel size=2.5 mm
3, four unweighted scans, and 30 diffusion-weighted scans at b=900 s/mm
2; 2011 and 2015: eight unweighted scans and 60 diffusion-weighted scans at b=900 s/mm
2). The same head coil was used at both time points. Full acquisition details have been described previously (
11).
Brain Volumetry
Gray matter volume, white matter volume, and CSF volume were assessed using SPM12 (
https://www.fil.ion.ucl.ac.uk/spm) unified segmentation routines on the T
1 MPRAGE images corrected for WMHs, as described in detail previously (
14). Intracranial volume (ICV) was calculated by summing gray matter volume, white matter volume, and CSF volume as well as total brain volume by summing gray matter and white matter volumes. To correct for interscan effects, we corrected for differences in ICV between baseline and follow-up. We normalized all volumes to baseline ICV to account for head size. Hippocampal volume segmentations were automatically processed with the longitudinal stream in FreeSurfer, version 5.3 (
15). The calculation of hippocampal segmentations has been reported in detail previously (
16).
MRI Markers of SVD
MRI markers of SVD were rated according to the Standards for Reporting Vascular Changes on Neuroimaging (STRIVE) criteria (
3). WMH volumes were calculated by a semiautomatic WMH segmentation method, described in detail elsewhere (
17). Segmentations were visually checked for segmentation errors by one trained rater, blinded for clinical data. To minimize the effects of changes in FLAIR acquisition sequence parameters, follow-up FLAIR images were resliced to match baseline slice thickness using FMRIB’s Linear Image Registration Tool (FLIRT), which is part of FSL. WMH volumes were normalized to ICV. Follow-up WMH volumes were corrected to baseline ICV, so that WMH volumetric changes over time could be quantified. Lacunes and cerebral microbleeds were rated manually on FLAIR/T
1-weighted and T
2*-weighted MRI scans, respectively. Both markers were rated by two trained raters blinded for clinical data, with excellent interrater agreement in a random sample of 10% of the scans (weighted kappa values were 0.87 and 0.95, respectively, for the number of lacunes, and 0.85 and 0.86, respectively, for the number of cerebral microbleeds) (
18).
To facilitate consistent identification of incident lacunes, difference images were constructed for T
1 and FLAIR images to identify incident lacunes. The images were first fully stripped using the Brain Extraction Tool (BET) in FSL. All follow-up images were then registered to the baseline scans. Difference images were generated by subtracting the registered and intensity-normalized baseline T
1 and FLAIR images from the corresponding T
1 and FLAIR images at the follow-ups. Incident lacunes were defined as hypointense voxel clusters on a uniform background (
19).
Diffusion Tensor Imaging Preprocessing
All diffusion-weighted images were denoised using a local principal component analysis filter and corrected for cardiac, head motion, and eddy current artifacts simultaneously using the “PATCH” algorithm, as described previously (
20). Diffusion-weighted-imaging-positive (DWI+) lesions were rated on mean diffusivity and trace images. The peak width of skeletonized mean diffusivity (PSMD) was calculated with the PSMD tool provided at
http://www.psmd-marker.com (
21).
Dementia Diagnostic Workup
Dementia case finding was performed for all patients, with a different approach for those who participated in the in-person study visit than for those who died or declined in-person follow-up participation.
Patients who participated in the in-person follow-ups (in 2011, 2015, and 2020) were first screened with the Mini Mental State Examination (MMSE) (
22) and the Instrumental Activities of Daily Living (IADL) scale (
23) during all follow-up waves. Participants screened positive if they fulfilled two criteria: 1) an MMSE score below 26 or a decline of ≥3 points compared to the previous assessment, and 2) having an impairment on least one item of the IADL scale (i.e., a score <8). Patients who screened positive were subsequently evaluated in an independent multidisciplinary consensus panel meeting consisting of a neurologist, a clinical neuropsychologist, and a geriatrician. Adjudication of dementia diagnosis was done by evaluating all available clinical data, neuropsychological examinations, depressive symptom scores (assessed with the Center for Epidemiologic Studies Depression Scale [CES-D]) (
24), and MRI data from all time points available. In addition, for every patient who screened positive, the general practitioner or nursing home treating physician as well as family members or other informal caregivers were contacted to provide a comprehensive history and evaluation of the interference of cognitive deficits in the patient’s daily functioning. In cases where there was a need for an additional in-person evaluation, patients were subsequently referred to the Radboudumc Alzheimer Center memory clinic.
For patients who did not participate at the follow-up time points (i.e., those who were deceased or who declined participation), medical records on their cognitive status were retrieved from the general practitioner or nursing home treating physicians. Cases of patients diagnosed with dementia or a cognitive disorder by their own treating physician were reviewed again independently by the members of the consensus panel, additionally taking into consideration the already collected research data on cognitive function and mood, activities of daily living, decline over time during previous visits, and all available neuroimaging.
The diagnosis of dementia was based on the DSM-5 clinical criteria for major neurocognitive disorder due to a neurodegenerative disease. The disease criteria for Alzheimer’s dementia were based on the National Institute on Aging and Alzheimer’s Association (NIA-AA) Research Framework (
25); our patients in this dementia category were described as having Alzheimer’s clinical syndrome. Vascular dementia was specified using the National Institute of Neurological Disorders and Stroke and Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINDS-AIREN) disease criteria (
26). Individuals with a dementia diagnosis who did not meet these criteria were classified as having etiologically mixed dementia (mixed Alzheimer’s dementia/vascular dementia) (
27), frontotemporal dementia, or Parkinson’s dementia.
The date of dementia onset was defined as the date of diagnosis. When the date of diagnosis was not exactly known (for example, when diagnosed at the consensus meeting), we used the midpoint between the date the diagnosis was confirmed and the previous visit. Participants who did not develop dementia were censored at the time of last contact, date of most recently retrieved medical information regarding cognitive status from general practitioners, or death.
Statistical Analysis
Differences in baseline characteristics between subgroups were compared using Mann-Whitney U tests, one-way analysis of variance, or chi-square tests as appropriate. WMH volumes were log-transformed to achieve a normal distribution.
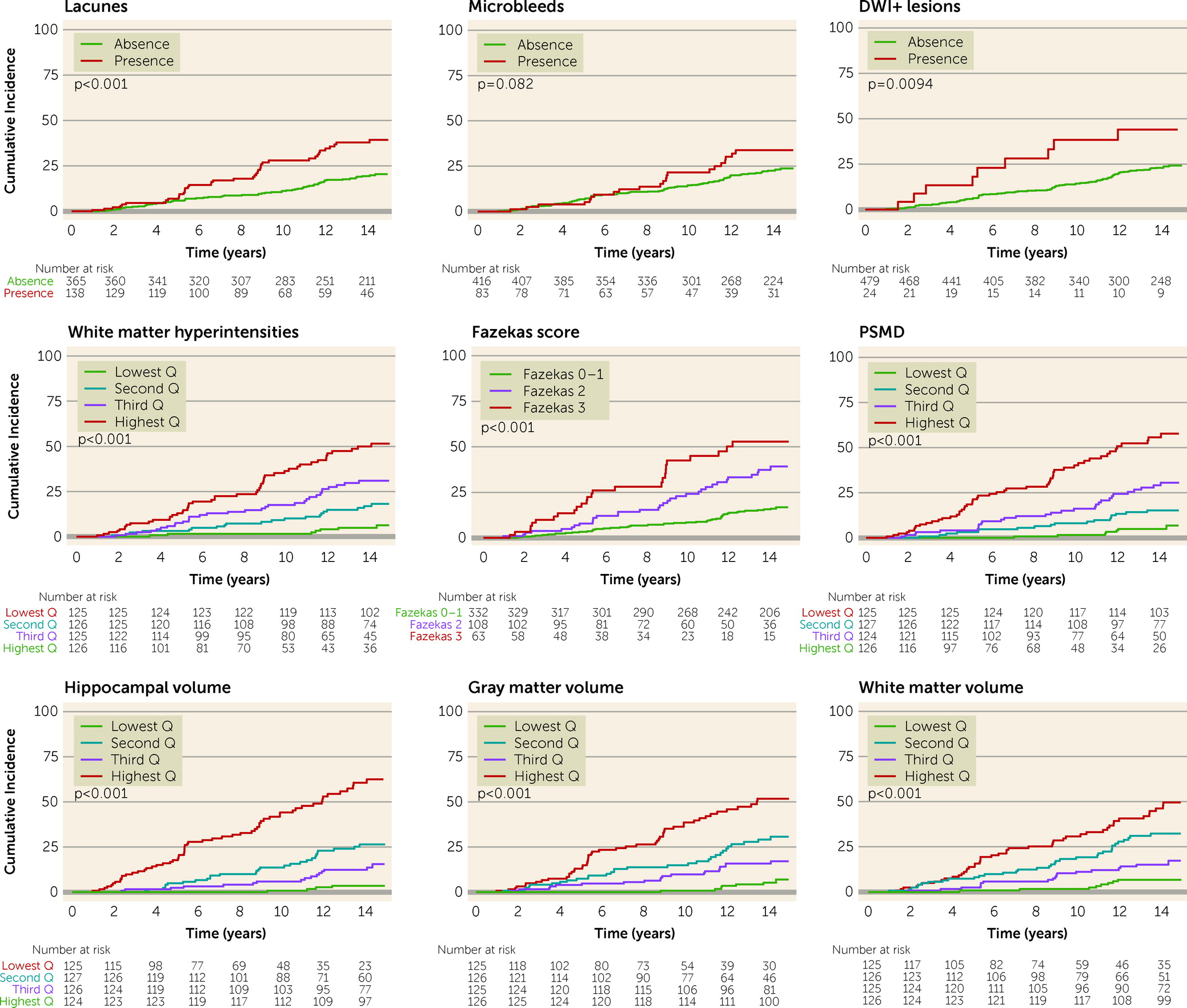
Person-years at risk was calculated from date of baseline assessment until onset of dementia, death, or date of last follow-up. Cumulative incidence of all-cause dementia was calculated by using Kaplan-Meier analyses for the different MRI markers of SVD, stratified by severity. Differences were tested using log-rank tests.
Cox proportional regression analyses were used to study the association between MRI markers of SVD and dementia incidence. Two models were constructed: in the first model, adjustments were made for age, sex, CES-D score, education level, and the vascular risk factor score (based on the number of vascular risk factors [0–4] at baseline). In the second model, additional adjustment was made for hippocampal volume as an imaging marker for concurrent neurodegenerative processes. Conversely, to study hippocampal volume in relation to dementia incidence, adjustment was made for PSMD (as this is considered to be the most sensitive marker capturing SVD) (
21). To eliminate the effect of unintentionally included subclinical dementia cases at baseline, sensitivity analyses were performed by excluding participants who developed incident dementia in the first 5 years, until the first follow-up (2011). We performed similar Cox regression analyses between baseline MRI markers of SVD and dementia after 2011. Death was considered a competing risk, by using the proportional hazards model of Fine and Gray (
28). Schoenfeld residuals were investigated to verify the proportionality of hazards. There were no indications that the proportional hazards assumption was violated. Similar sensitivity analyses were also performed specifically for Alzheimer’s dementia and vascular dementia.
The relation between SVD and dementia was further investigated by analyzing progression of MRI markers as determinants of incident dementia. Only patients who underwent at least two MRI scans between the follow-up assessments of 2006, 2011, and 2015 were included in these analyses. We included only dementia cases that occurred after a second MRI, to ensure use of serial MRI data prior to dementia conversion. We constructed linear mixed-effects models to estimate annualized rate changes of MRI markers, namely, WMH volumes, hippocampal volume, total brain volume, gray matter volume, and white matter volume. The intercept and slope of each participant’s linear trajectory were allowed to vary with both fixed and random effects. Fixed-effect variation was accounted for by time, and random-effect variation allowed for interindividual differences. Slopes for each patient were extracted and used for further analyses.
Three models were employed by using the Fine and Gray hazards model and taking death as a competing risk: in model 1, the progression of MRI markers was adjusted for age, sex, education level, and risk factor score; in model 2, each marker was additionally adjusted for its baseline measure; and in model 3 we additionally adjusted for baseline hippocampal volume; hippocampal volume was adjusted for baseline PSMD.
All statistical analyses were performed in R, version 4.1.1.
Discussion
We found that SVD severity at baseline and SVD progression over a 9-year course were independently associated with an increased risk of incident all-cause dementia over a follow-up of 14 years. These observations provide strong starting evidence for a causal role of SVD in the development of dementia.
Our results regarding long-term dementia incidence extend the existing knowledge of short-term dementia incidence in SVD. Higher dementia incidences were reported for previous SVD cohorts with shorter follow-ups—18.2% in the St George’s Cognition and Neuroimaging in Stroke (SCANS) study (
29) and 14.0% in the Leukoaraiosis and Disability (LADIS) study (
30) after a median follow-up of approximately 3 years—compared with the incidence of 21.5% we observed over 14 years of follow-up. However, the patients included in these studies were much older and had higher cardiovascular and SVD burden at baseline compared with our cohort, which is a more representative sample of community-dwelling individuals.
Our finding that SVD severity at baseline is associated with an increase in risk of dementia is in line with previous studies (
4,
31–
33). Yet, the majority of these studies had relatively short follow-ups, of only several years, and did not adjust for hippocampal volume (
4,
7,
31). It may therefore be that ongoing neurodegeneration explained the development of dementia soon after baseline and that the patients included in those shorter studies already had (subclinical) signs of cognitive impairment at baseline. This issue is countered by following patients over a longer follow-up.
Higher WMH volume, presence of DWI+ lesions, and higher PSMD were independent SVD predictors for all-cause dementia. PSMD has previously been shown to outperform conventional MRI markers of SVD (e.g., WMHs) when studying SVD severity in relation to cognitive impairment (
21) and is considered the most reliable MRI marker capturing SVD. Our findings here clearly show that it can also predict long-term all-cause dementia. Detection of DWI+ lesions also independently predicted all-cause dementia after a follow-up of 14 years. This extends the findings of a previous study conducted in memory clinic patients showing that DWI+ lesions were associated with an increased risk of vascular dementia (but not all-cause dementia and not with adjustment for hippocampal volume) after a median follow-up of 2 years (
5). Therefore, despite its low occurrence, presence of DWI+ lesions may serve as a marker of “active” SVD reflecting further ongoing vascular brain damage, and may thus guide targeted prevention strategies by identifying high-risk patients.
We noted that presence of most MRI markers of SVD was associated with a higher risk of incident vascular dementia, yet a lower risk of Alzheimer’s dementia. This seems counterintuitive, since SVD is increasingly recognized in the etiology of Alzheimer’s dementia (
34). However, this can be explained by confirmation bias during our diagnostic workup, as the presence of severe SVD favors a vascular dementia diagnosis on the basis of accepted diagnostic criteria (
26), resulting in a few Alzheimer’s dementia cases with severe SVD burden.
Given our longitudinal design and long follow-up with multiple cognitive and MRI evaluations, we were able to study the 9-year SVD progression on incident all-cause dementia. WMH progression and incident lacunes (however, not after adjustment for hippocampal volume) were significantly associated with incident all-cause dementia, as well as hippocampal, gray matter, and white matter atrophy. Of note, the effect of progression of these MRI markers on dementia in our study is most likely an underestimation, since attrition bias may have occurred: older participants with higher SVD burden at baseline may not have been able to participate in the follow-up assessments, either because they were deceased or because they had greater frailty. We previously demonstrated (
35) that mainly these patients with high SVD burden also show the highest SVD progression rate over time, and therefore the loss of these patients to follow-up may have attenuated the true effect of SVD progression on dementia.
Our results extend previous smaller longitudinal studies investigating SVD progression over a much shorter follow-up and with varying populations in terms of age, cognitive status, and cardiovascular disease burden. In one previous study, WMH progression and deterioration of microstructural integrity within 3 years were found to be related to dementia incidence (
7). However, that study had smaller numbers of patients with incident dementia (N=18) and included only patients with lacunar stroke who were much older and had higher SVD burden. In another pair of studies, 4-year WMH progression was associated with cognitive decline in both cognitively unimpaired older individuals and patients with Alzheimer’s dementia (
36,
37). However, these studies did not adjust for any neurodegenerative markers.
Equivocal results have contributed to a long debate on whether and how SVD relates to dementia. It is proposed that SVD causes cognitive decline by directly disconnecting white matter pathways underlying complex cortical-subcortical networks involved in efficient processing of cognitive and memory functions (
16), or via synergistic interaction with Alzheimer’s pathology, that is, hippocampal atrophy and β-amyloid load (
16,
38). Another study showed a causal relation between incident subcortical infarcts and atrophy of connected cortical regions (
39). However, other alternative mechanisms, including overlapping genetic risk profiles, inflammation, and a disrupted cholinergic system, have also been suggested to interact in this relation (
40,
41). Since depression is associated with presence of SVD and incident dementia, we adjusted for depressive symptoms in our multivariable analyses. Still, the MRI markers were associated with incident dementia independent of depressive symptoms.
SVD has cross-sectionally been related to hippocampal atrophy, but other concurrent neurodegenerative processes likely affect hippocampal volume as well. Yet, by adjusting for hippocampal volume, we may have partially adjusted for the effect of SVD, even though hippocampal volume is not an established MRI marker of SVD. Of note, we showed that even when we adjusted for hippocampal atrophy, SVD severity at baseline and its progression over time were still significantly associated with dementia incidence.
Some limitations of our study need to be considered. Even though the dementia diagnosis adjudication was done in a structured and standardized way, in which a consensus panel consisting of experts from different fields reviewed all available clinical, cognitive, and MRI data (including SVD severity and hippocampal atrophy), the diagnosis of Alzheimer’s dementia did not involve identification of β-amyloid or tau levels, or genetic profiles, which could improve the accuracy of Alzheimer’s dementia diagnosis. However, this only limits the stratified analyses and does not apply to all-cause dementia incidence, since this is often a clinical diagnosis. Another limitation is that the status of the large arteries was not evaluated during the study, and therefore we cannot preclude the possibility that the MRI markers of SVD could to some extent be caused by hypoperfusion or cerebral emboli resulting from carotid stenoses or intracranial stenoses, which may also mediate the relation between WMHs and cognitive decline (
42,
43). Nevertheless, this does not influence the clinical implication and interpretation of finding an association between presence of the studied MRI markers (regardless of their etiology) and incident dementia. Besides, the risk of misclassifying large artery disease as SVD seems small, given the low prevalence (<2%) of carotid or intracranial stenosis (according to the TOAST criteria) in asymptomatic White individuals (
42,
44).
Furthermore, there was a scanner change between 2006 and 2011, which may have led to measurement variability due to differences in slice thickness and FLAIR contrast. Consequently, we were not able to study longitudinal changes in diffusion MRI measures. However, these differences are almost inevitable with improvements in hardware and sequence design, especially in very long follow-up studies. Nevertheless, we minimized WMH measurement differences by reslicing the follow-up FLAIR scans to match baseline scans.
There are several strengths of this study. We present a long follow-up period (14 years) in SVD patients, with data acquired from a comprehensive and standardized clinical and neuroimaging workup, and with very few participants lost to follow-up (1.0%). Such a long follow-up offers insights into the long-term dementia outcome of SVD during a clinically relevant time period. For example, this long-term follow-up revealed that the largest differences in the cumulative risk of dementia by SVD severity only become apparent after 10 years, and not within several years after baseline, an interval chosen in most follow-up studies investigating SVD outcomes. Another strength of this study is the ability to investigate the (annual) 9-year progression of SVD markers in relation to incident dementia. Furthermore, in contrast to many other studies, we adjusted for hippocampal volume, an important neurodegenerative confounder in incident dementia, thereby increasing the clinical significance of the SVD markers.
In summary, both baseline SVD severity and SVD progression are independently associated with the long-term risk of all-cause dementia. Our results suggest that SVD progression precedes dementia and may causally contribute to its development, and consequently position SVD as a modifiable target for dementia prevention. Intensive blood pressure management has been shown in one trial to reduce SVD progression (
45), although another large trial recorded no effect of intensive cardiovascular management on all-cause dementia incidence (
46), indicating that therapeutic options for SVD should extend beyond adequate management of cardiovascular risk factors. Agents improving endothelial dysfunction, blood-brain barrier integrity, and anti-inflammatory effects, but also preventive strategies for managing vascular risk factors earlier in life, have been proposed as potentially promising therapeutic targets for SVD and thus dementia (
47). A delay in disease onset by merely a few years could reduce the devastating burden of dementia substantially.