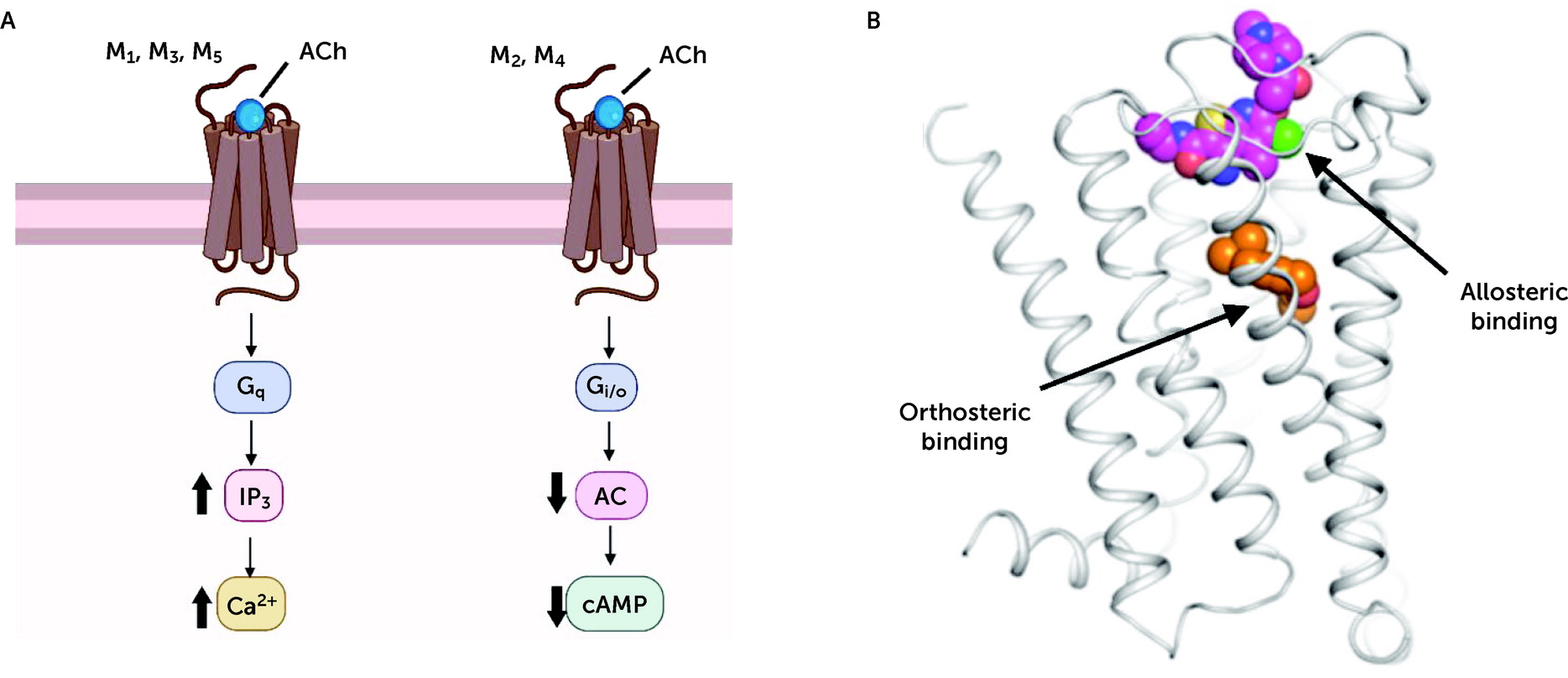
Xanomeline has been reported to bind with relatively equal affinity to all five mAChRs, as determined by the displacement of the nonselective radiolabeled mAChR antagonist [
3H]-
N-methyl scopolamine (
84,
85), and has been reported in some (
86) but not all (
26,
85) studies to bind to several serotonin (5-HT) receptors with low to high nanomolar affinity. Historically, radioligand binding assays with receptor-containing isolated whole-cell or synaptic membranes were used to rapidly screen compounds that target GPCRs, but these simple binding assays do not adequately establish whether a given compound of interest is an agonist or an antagonist or, importantly, whether it has functional activity as either a full or partial agonist or antagonist (
42). Efforts have been made to develop signaling-dependent cell-based functional assays to provide more accurate and comprehensive data sets of compounds targeting GPCRs. Based on several cellular and in vivo functional assays, xanomeline appears to be selective for M
1 and M
4 receptors, and at pharmacologically relevant concentrations, it does not have significant functional activity at other GPCRs (
85,
87,
88), including DA or 5-HT receptors.
Recent studies using X-ray crystal or cryo-electron-microscopy–solved structures of mAChRs at the <3.0-angstrom level, along with in silico molecular dynamic simulations, have allowed more detailed descriptions of both the static and kinetic interactions of mAChR ligands, such as xanomeline, at each of the mAChR subtypes (
89). Based on these studies, xanomeline has been found to have unique and unexpected binding properties and pharmacology, including functional selectivity for M
1 and M
4 receptors (
24,
90) and some level of persistent or “wash-resistant” binding that suggests pseudo-irreversible binding properties (
91–
93), which may contribute to its potentially unique functional pharmacology. Data generated from pharmacological models (
94), paired with genetic deletion of mAChRs in mice (
49), have provided considerable insight into which receptor subtypes are responsible for xanomeline’s behavioral pharmacology.
Xanomeline exhibits robust “antipsychotic drug–like” activity in animals treated with psychostimulant drugs that increase DA neurotransmission (e.g., amphetamine or apomorphine) (
25,
26,
90,
95–
97) or that block
N-methyl-
d-aspartate (NMDA) receptors (e.g., phencyclidine [PCP], dizocilpine [MK-801], or ketamine) (
98,
99). The ability of xanomeline to decrease the functional effects of dopaminergic and glutamatergic psychostimulants is consistent with the putative antipsychotic actions of this compound, findings that have been replicated across numerous preclinical behavioral models of “psychosis” (
24,
25,
90,
95,
97,
98,
100) and in recent pharmacological MRI studies (
101,
102). Moreover, xanomeline’s antipsychotic activity is fully blocked by a centrally penetrant but not peripherally restricted pan-muscarinic-subtype antagonist, indicating that activation of one or more central mAChRs is sufficient to elicit antipsychotic-like activity (
103) and likely to regulate neurotransmitter systems and neural circuits implicated in the pathophysiology of schizophrenia. Evidence for which mAChR subtypes are involved in the behavioral actions of xanomeline has only recently been made possible with the development of mice bearing genetic deletions of one or more mAChR subtypes as well as by using mAChR subtype–selective agonists.
Role of M4 Receptors
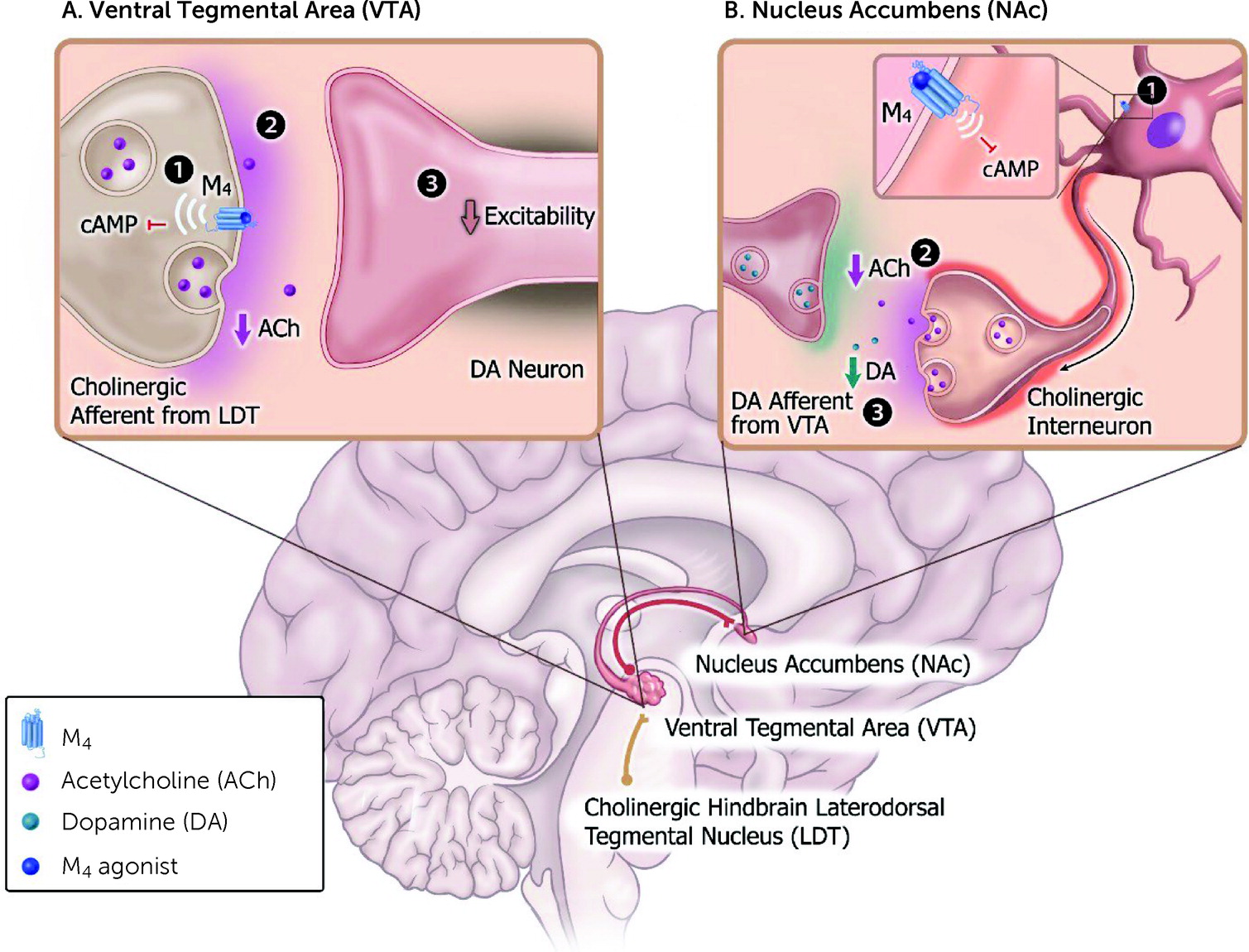
The onset of psychotic (i.e., positive) symptoms is believed to be associated with neural network dysfunction that includes a variety of brain regions and neurotransmitters, including increased activity of ventral tegmental area (VTA) DA neurons and heightened terminal DA release (
104). Although the exact mechanism of how antipsychotic drugs achieve their therapeutic activity is still not completely understood, it has been postulated, with considerable experimental support, that most, if not all, antipsychotic drugs induce “depolarization block” or inactivation of DA neurons in both the VTA (“mesolimbic DA circuit”) and the substantia nigra (SN; “motor DA circuit”); this phenomenon takes place over a period of time that parallels the delay in the onset of clinical improvement (
105). Based on the “depolarization block” hypothesis, antipsychotic drugs with more selective actions on VTA DA neurons consistently show a lower motor symptom liability (i.e., low extrapyramidal side effects), whereas high liability to extrapyramidal side effects is associated with antipsychotic drugs that more markedly reduce the activity of DA neurons in the SN, which is involved in the control of the extrapyramidal motor control system (
106,
107). Importantly and often overlooked is that the activity of midbrain VTA DA neurons is modulated by cholinergic input from hindbrain structures (
108), particularly the laterodorsal tegmental nucleus (LDT), where M
4 receptors are abundantly expressed and function as autoreceptors (
49,
109). M
4 receptors play a key role in blunting the stimulatory effects of ACh on DA neurons, making them less active and thereby reducing downstream DA release in brain regions implicated in psychosis, such as the nucleus accumbens and ventral striatum (
Figure 5A) (
49,
109).
Interestingly, xanomeline has been shown to selectively and rapidly reduce VTA DA neuron firing rates but not SN DA neuron activity (
24), an effect now believed to be mediated by selective activation of LDT afferents (
110,
111). This very rapid reduction in VTA DA neuron activity observed following acute administration of xanomeline (
24) contrasts with the slow, time-dependent depolarization block of these same DA neurons that occurs following treatment with second-generation antipsychotics (
105,
112,
113). The DA D
2 receptor–independent modulation of DA microcircuits may also explain the relatively rapid antipsychotic effects of xanomeline reported in patients with Alzheimer’s disease (
22) or schizophrenia (
23). Also consistent with xanomeline’s selective action on mesolimbic VTA DA neurons is the lack of overt motor effects (e.g., catalepsy) observed in preclinical models (
24,
90) or its lack of effect in inducing immediate early gene expression (e.g., cFos) in brain regions associated with extrapyramidal side effects (
114). This highly selective regulation of DA-containing neural circuits seen with xanomeline may therefore account for the lack of observed extrapyramidal side effects in multiple clinical trials (
22,
23,
82) and a much lower, if not absent, risk of developing tardive dyskinesia.
In addition to their ability to regulate midbrain VTA DA neuron activity, M
4 receptors are expressed in cholinergic interneurons that reside locally in the nucleus accumbens and modulate terminal DA release (
115,
116). Within the nucleus accumbens, activation of M
4 autoreceptors on cholinergic interneurons decreases spontaneous activity, which reduces ACh release to dampen nicotinic receptor feed-forward stimulation on DA terminals (
Figure 5B) (
116,
117). It should be noted, however, that there are other postulated mechanisms by which M
4 receptor–mediated inhibition of local DA release may occur, such as activation of M
4 receptors coexpressed in D
1 receptor–expressing medium spiny neurons (MSNs) of the dorsolateral striatum (
118,
119). In support of this hypothesis, selective deletion of M
4 receptors from D
1 MSNs increases DA-dependent behavioral phenotypes (
119) and blunts M
4 receptor–mediated inhibition of DA release in preclinical models (
120), an effect likely mediated through competition of convergent second messenger systems (
56), recruitment of endocannabinoids (
120), interactions with G
q-coupled receptors (
121), or direct actions on enhanced Ca
2+ currents via Ca
v1 channels (
122). Genetically eliminating M
4 receptors from D
1 MSNs also markedly reduces the inhibitory effects of xanomeline on amphetamine-stimulated locomotor activity (
118).
More recent studies using highly selective M
4 receptor PAMs support the important role that these receptors play in mediating antipsychotic drug–like behavioral activity, as numerous chemical scaffolds that modulate M
4 receptors display antipsychotic activity (
60,
120,
123–
125). In addition to modulating classical neural circuits implicated in psychosis, M
4 receptor PAMs have been shown to enhance attentional and memory network function in preclinical rodent models (
126). M
4 receptor agonists can reduce elevated CA1 pyramidal neuron activity, which may partially contribute to their procognitive effects (
85), as changes in CA1 excitability have been postulated to contribute to the cognitive deficits in schizophrenia (
27). However, additional detailed studies are needed to fully understand the multinodal mechanisms by which M
4 receptors impact local subcortical microcircuits within the dorsal (i.e., associative striatum) and ventral striatum to inhibit DA neurotransmission (for a review, see reference
9).
Although M
4 receptor regulation of DA microcircuits appears to be important in the central mechanism of action of xanomeline, glutamatergic microcircuits may also contribute to its antipsychotic activity. In rodents, acute administration of NMDA receptor antagonists, such as PCP or MK-801, produces behavioral hyperactivity and cognitive deficits that correlate with a disinhibition of pyramidal cell firing in the prefrontal cortex and increased DA and glutamate levels (
127,
128). Administration of xanomeline or a selective M
4 receptor PAM can attenuate PCP- or MK-801-evoked locomotor activity, an effect that is absent in global M
4 receptor knockout mice (
123). M
4 receptor–mediated regulation of MK-801-induced hyperactivity may involve actions at corticostriatal terminals to normalize the function of overactive excitatory glutamatergic projections to the striatum (
129) or via a dampening of thalamocortical synapses (
130). This “top-down” control of cortical glutamatergic projections has important implications, as these projections indirectly modulate phasic DA release (
118). In addition, activation of M
4 receptors attenuates MK-801-induced disruptions in learning and memory (
123) and elevations of high-frequency gamma power as well as state-dependent alterations in sleep architecture and arousal similar to the effects observed with atypical antipsychotics in preclinical models (
131). Taken together, these findings suggest an important role for M
4 receptors in modulating neural circuits involved in the psychotic, motivational, cognitive, and executive functions disrupted in schizophrenia via glutamatergic microcircuits.
These behavioral studies describing an important role for M
4 receptors in animal models of psychosis have led to further studies exploring the potential role of M
4 receptors in the pathophysiology of schizophrenia. In postmortem studies, M
4 receptor expression has been reported to be decreased in striatal and hippocampal brain regions of patients with schizophrenia compared with those of healthy control subjects (
132). Other studies using modestly selective mAChR antagonist radioligands also suggest that M
4 receptors are decreased in the frontal cortex, hippocampus, and striatum of patients with schizophrenia compared with those of control subjects (
133,
134). In tandem with the apparent decreases in M
4 receptor expression, these morphometric changes could also contribute to decreased regional brain volumes and cortical thickness. Genetic markers (single-nucleotide polymorphisms) in a region on chromosome 11 have been reported to be associated with schizophrenia (
135), and this region contains several candidate genes, including
CHRM4, the gene encoding the M
4 receptor. Two genomic variants of the M
4 receptor, rs2067482 and rs72910092, have also been reported to be associated with an increased risk of schizophrenia (
136). It should be noted, however, that these allelic associations have not been consistently replicated across studies (
137), which may be due to differences in the patient populations sampled and diagnostic subtyping.
Importantly, recent clinical results were released from a phase 1b placebo-controlled trial of emraclidine (CVL-231), an M
4 receptor PAM, in patients with schizophrenia (
138). In an exploratory analysis, treatment with CVL-231 was associated with reduced psychotic symptoms compared with placebo after 6 weeks of treatment (
138). Rates of gastrointestinal adverse events were minimal compared to those in historical mAChR agonist trials (
41,
69). These data provide additional evidence that M
4 receptors play key roles in mediating the antipsychotic properties of mAChR agonists (
138) and preliminary clinical validation for drugs that target allosteric binding pockets of mAChR receptors (in this case M
4 receptors). Although encouraging, these data have yet to be peer reviewed, and further placebo-controlled trials of CVL-231 will be necessary to establish its efficacy and safety profile in patients with schizophrenia.
Role of M1 Receptors
Although an important role for M
4 receptors in mediating the antipsychotic effects of xanomeline is quite likely (see above) there is also substantial evidence that M
1 receptors regulate neural circuits underlying psychosis as well as learning and memory (especially working memory). Xanomeline’s “antipsychotic” activity as measured by amphetamine-induced hyperactivity was abolished in mice lacking M
4 receptors but also was partially attenuated in mice lacking M
1 receptors, suggesting that M
4 receptors and, to a certain degree, M
1 receptors may both contribute to xanomeline’s efficacy in treating the positive symptoms of schizophrenia (
97). As xanomeline is a “dual” M
1 and M
4 receptor agonist, M
1 receptors may also contribute to its reported antipsychotic and procognitive activity.
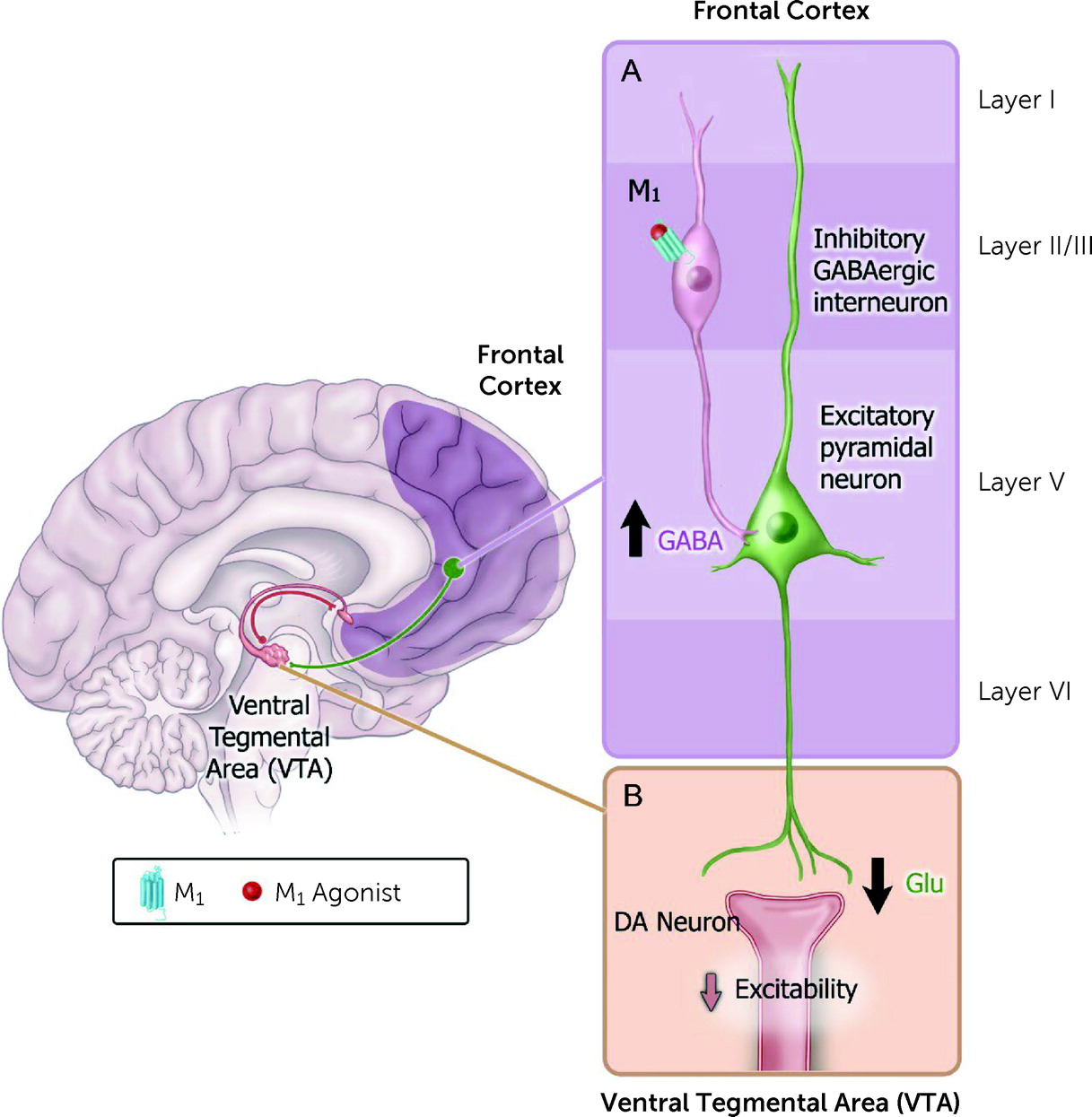
Several hypotheses have emerged regarding how M
1 receptors may modulate neural circuits implicated in psychosis, including regulation of top-down circuits that synapse onto VTA DA neurons (
Figure 6) (
139,
140). For example, activation of M
1 receptors facilitates excitability of cortical GABAergic interneurons that synapse onto pyramidal neurons (
139), causing a decrease in excitability of principal cortical output neurons. However, there are additional hypotheses regarding how M
1 receptor activators may exhibit antipsychotic activity, including augmentation of corticostriatal plasticity (
141), modulation of MSN excitability (
142), and enhanced communication between MSNs (e.g., via nucleus accumbens output neurons) (
143). Additional studies have demonstrated a “psychosis-like” phenotype in global M
1 receptor knockout mice (
144) as well as antipsychotic drug–like activities in various preclinical models following administration of selective M
1 receptor PAMs (
145,
146). M
1 receptor PAMs have also been reported to reverse excessive spontaneous locomotor activity in NMDA receptor NR1-subunit knockdown mice that display an NMDA receptor–mediated hypofunction phenotype (
147).
As summarized above, significant effort has been made to develop M
1 receptor agonists to treat the cognitive impairment associated with various neuropsychiatric and neurodegenerative disorders (
64,
70). In memory circuits, M
1 receptor activators have been shown to enhance synaptic plasticity (
85,
148), increase neuronal excitability (
149), and facilitate learning and improve cognition in aged animals (
150) and in a variety of NMDA receptor hypofunction models of impaired learning and memory (
147,
149,
151,
152). Previous studies have shown that M
1 receptors are physically and functionally coupled to NMDA receptors and that activation of M
1 receptors potentiates NMDA receptor currents in cortical and hippocampal pyramidal neurons (
54,
153,
154). Conversely, global M
1 receptor knockout mice have impaired performance in prefrontal cortex–dependent cognitive tasks (
155,
156) and reduced hippocampal long-term potentiation (
157). Administration of an M
1 receptor potentiator in genetic models of NMDA receptor hypofunction restored plasticity deficits and improved impaired learning and memory in these mice (
147). Moreover, M
1 receptor PAMs have been shown to modulate sleep-wake architecture in rodents and nonhuman primates (
150), suggesting that activation of M
1 receptors may participate in restorative sleep–mediated plasticity, which has been postulated to be dysregulated in patients with schizophrenia (
158). Beyond cognition, M
1 receptor PAMs have been reported to reverse behavioral and electrophysiological deficits in chronic PCP rodent models, which are thought to recapitulate the “deficit state” or negative symptoms of schizophrenia (
149). Recently it was shown that M
1 receptor PAMs can reverse PCP-induced disruption of mAChR-stimulated long-term depression (
149), a plasticity measure that is important for adapting neural networks to physiological activity. These studies support the idea that M
1 receptor activation may, by itself, contribute to improvements in the cognitive, negative, and even positive symptom domains of schizophrenia, and thus a dual M
1/M
4-preferring mAChR agonist may be particularly effective in treating schizophrenia.
As mentioned earlier, clozapine appears to be unique among the drugs used to treat schizophrenia, as it appears to be more effective in treating positive and negative symptoms in patients with treatment-resistant illness (
159). Moreover, some (
160) but not all (
161) studies have reported that clozapine can improve cognition and especially working memory performance in patients with schizophrenia. Interestingly, although clozapine itself is a potent mAChR antagonist (
162), its major metabolite,
N-desmethylclozapine (NDMC), is a potent partial M
1 receptor agonist (
163). Recently, it has been reported by several groups that lower clozapine-to-NDMC ratios are associated with improvements in working memory and executive function, whereas higher ratios are associated with cognitive deficits (
164–
167). These findings raise the intriguing possibility that the M
1 receptor activity of NDMC contributes to clozapine’s unique clinical profile, including its reported procognitive benefits (
160). Several lines of evidence suggest that M
1 receptors may also play a key role in the ability of clozapine to modulate schizophrenia-related circuity in rodents (
168–
170). Sub-efficacious doses of clozapine can reverse MK-801-induced deficits in sensorimotor gating, and this is potentiated by coadministration of an M
1 receptor PAM (
151). From a circuit-level perspective, local administration of NDMC can alter DA release in brain regions implicated in psychosis, an effect that is opposite to that of clozapine (
171).
However, in preclinical models (
172), and in marked contrast to clozapine, NDMC did not display antipsychotic activity, and a subsequent phase 2 clinical trial in patients with schizophrenia confirmed its lack of efficacy in treating positive symptoms (
173). Although NDMC shares many of clozapine’s pharmacological properties beyond mAChR activity, it does not occupy or block DA D
2 receptors (
174); this may account for its lack of efficacy in treating positive symptoms. However, it is quite possible that NDMC accounts for the procognitive effects of clozapine and possibly its beneficial effects on negative symptoms. NDMC could also be responsible for some of the peripheral adverse effects observed with clozapine (
175), such as hypersalivation. Moreover, the peripheral gastrointestinal adverse events reported for xanomeline are almost certainly due to stimulation of peripheral mAChRs. Nonetheless, these data fit with the hypothesis that M
1 receptor activation could be a key mechanism through which clozapine exerts its unique clinical profile (
175).
Previous studies have also suggested a role for M
1 receptors in the pathophysiology of schizophrenia. For instance, using [
123I]quinuclidinyl benzilate, mAChR availability was found to be reduced in unmedicated patients with schizophrenia (
21), a finding similar to that in the postmortem studies. Several postmortem studies using the mAChR antagonist [
3H]-pirenzepine have demonstrated decreased M
1 receptor expression in cortical and subcortical regions in patients with schizophrenia (
62,
176,
177). The authors refer to these patients as having “mAChR deficit” schizophrenia. Interestingly, whereas there is a pronounced loss of M
1 receptors in the mAChR deficit schizophrenia subtype, the residual M
1 population has increased receptor–G protein coupling efficiency, suggesting an adaptive change to compensate for reduced receptor expression (
178). Importantly, these changes in M
1 receptor expression appear to be specific to schizophrenia and may represent a distinguishable endophenotype (
177). Additional evidence that M
1 receptors may contribute to the pathophysiology of schizophrenia include recent reports that homozygous carriers of
CHRM1 C267A nucleotide polymorphisms exhibit pronounced perseveration errors and poor performance on tests of executive functioning (
179,
180). More recently, elevated serum titers of anti–M
1 receptor antibodies have been reported in up to one-third of people diagnosed with schizophrenia (
181,
182), and their presence was correlated with the severity of negative symptoms (
181).
In actual practice, it is likely that many patients with schizophrenia will be treated simultaneously with conventional antipsychotic drugs and novel therapies to improve overall efficacy before they are switched to monotherapy with a novel drug (
183). This concept is supported by preclinical data showing that treatment with the M
1 PAM BQCA in combination with atypical antipsychotics (i.e., aripiprazole and clozapine) provided synergistic procognitive activity in deficit states induced by the NMDA receptor antagonist MK-801 (
151). Although additional studies are needed to fully understand the underlying neural circuits involved in mediating the procognitive effects of BQCA, they likely involve modulation of hippocampal synaptic plasticity (
184,
185).
In the context of psychosis, subeffective doses of the M
4 PAM PGM039678 in combination with subeffective doses of the atypical antipsychotics olanzapine or risperidone significantly augmented conditioned avoidance responding in rats (
186). Recently, it was also reported that subeffective doses of xanomeline augmented the activity of aripiprazole and risperidone in the conditioned avoidance response assay in mice (
103). In the same study, subthreshold doses of xanomeline and risperidone administered together significantly attenuated MK-801-induced hyperactivity (
103). These results raise the intriguing possibility that M
1 receptor, M
4 receptor, or dual M
1/M
4 receptor agonists may represent adjunctive treatments, when used together with currently prescribed first- or second-generation antipsychotics, to improve the core symptoms of schizophrenia, especially in patients with treatment-resistant illness.
Role of M2, M3, and M5 Receptors
Pharmacological studies using M
2 receptor–preferring antagonists have produced contradictory results regarding their potential role in cognition. Whereas some studies suggest that blockade of central M
2 receptors enhances learning and memory in various experimental settings (
187), other studies arrived at the opposite conclusion (
188). M
2 receptor global knockout mice display deficits in behavioral flexibility, working memory, and passive avoidance learning (
189). However, M
2 receptors are apparently not required for stimulus-reward learning. Reduced expression of subcortical (
190,
191) but not cortical (
192) M
2 receptors has also been found in patients with schizophrenia. In preclinical models, the antipsychotic-like activity of BuTAC, an M
2 receptor–preferring orthosteric agonist and a partial M
1/M
4 receptor agonist, is lost in global M
2/M
4 double-knockout mice (
193). From a microcircuit perspective, there appears to be a functional interplay between M
2 and M
4 receptors to regulate DA release in striatal regions (
116), suggesting that M
2 receptor activation may play an important role in M
4-mediated suppression of DA release. Additional studies are needed to understand the therapeutic potential of dual M
2-M
4 receptor agonists or whether M
2-M
4 heterodimers play a role in psychosis.
The M
3 receptor is expressed widely in the CNS, including in the hippocampus (
194), and may play an important role in learning and memory. M
3 receptor knockout mice have severe deficits in hippocampus-dependent memory, suggesting that selective M
3 receptor activators may be beneficial for cognition (
195). Recently, it has been reported that M
3 receptors regulate feed-forward inhibition, which may facilitate memory consolidation by reducing interference signals (
196). Detailed biochemical studies support an important role for M
3 receptor signaling through phosphorylation events as contributing to these procognitive effects (
195) and raise the possibility that M
3 receptor–biased ligands that increase β-arrestin-dependent (non–G protein) signaling might promote cholinergic-mediated learning and memory (
197).
Finally, M
5 receptors may also be a promising drug target for schizophrenia given their expression in and control of midbrain DA neurons (
198). M
5 receptor global knockout mice display reduced striatal DA release and blunted responses in preclinical models of psychosis (
199). Activation of M
5 receptors increases the activity of midbrain DA neurons (
200) to facilitate terminal DA release (
201), suggesting that therapeutic agents that inhibit the M
5 receptor will decrease elevated DA levels reported in various forebrain regions in schizophrenia. More recent preclinical evidence suggests that M
5 receptor agonists or PAMs may also be effective for treating negative symptoms (
202) and cognitive deficits (
203). Continued efforts are needed to develop CNS-penetrant compounds with appreciable M
5 receptor agonist or antagonist selectivity to further elucidate their potential benefits (
204,
205). In this regard, xanomeline has been shown to be an M
5 receptor partial agonist (
206) and thus is likely to have a predominant M
5 receptor antagonist pharmacological profile (
206). The contribution, if any, of xanomeline’s M
5 receptor activity, along with its M
1 and M
4 receptor pharmacology, to its reported clinical (antipsychotic and procognitive) activity in patients with schizophrenia is unclear but cannot be excluded.