Major depressive disorder is the most prevalent mental health condition (
1), affecting 1 in 15 adults in the United States (
2), and it is the psychiatric diagnosis most commonly associated with suicide (
1,
3). The incidence of attempted suicide is about 20-fold higher in patients with major depression compared with the general population (
4). The Centers for Disease Control and Prevention estimated that over 44,000 individuals in the United States died by suicide in 2015; over 1 million persons reported having made a suicide attempt in the previous 12 months, and over 2 million adults reported having considered suicide in the previous 12 months (
5,
6).
Suicidal ideation is a major risk factor for suicide in patients with major depression (
7,
8). The interval between the onset of suicidal ideation and suicide attempt is often very short (
9), highlighting the need for urgent intervention and development of novel antidepressant therapies with a rapid onset. While standard antidepressant medications have demonstrated efficacy in the treatment of mood symptoms, including suicidal ideation (
10), they require 4–6 weeks for optimal effect. No medication has been approved for the emergency treatment of patients with major depression assessed to be at imminent risk for suicide (
11,
12).
Research on mood disorder pathophysiology has implicated abnormalities in glutamatergic transmission, along with histopathological changes in neural circuits that modulate emotional behavior (
13). This work contributed to the investigation of glutamatergic agents in treating major depression. Several small studies reported that the NMDA receptor antagonist ketamine improved depressive symptoms (
14–
19) and reduced suicidal ideation within hours of intravenous administration (
20–
22). Likewise, pooled analyses of placebo-controlled single-dose studies of ketamine (
23–
25) suggested a substantial reduction in suicidal ideation in patients with treatment-resistant or bipolar depression. While most of the participants in these studies were not specifically selected for the presence or severity of suicidal ideation, a recent study of 80 depressed patients with clinically significant suicidal ideation found that ketamine produced a greater reduction in suicidal ideation within 24 hours compared with midazolam (
26).
Esketamine, an NMDA receptor antagonist that modulates glutamatergic transmission (
27), is being developed as an intranasal formulation for treatment-resistant depression and for rapid reduction of symptoms of major depressive disorder, including suicidal ideation, in patients at imminent risk for suicide. Rapid onset of antidepressant effects has been observed in patients with treatment-resistant depression as early as 2 hours (
28) and 24 hours after single-dose intranasal esketamine administration (
29).
Here, we report findings from a proof-of-concept study conducted in the context of comprehensive standard-of-care treatment. To explore the hypothesis that esketamine is more efficacious than placebo, we compared standard-of-care treatment plus either intranasal esketamine or placebo for rapidly reducing depressive symptoms, including suicidality, among individuals with major depression who were assessed to be at imminent risk for suicide.
Method
Ethical Practices
Independent review boards approved the study protocol and its amendments. The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, consistent with Good Clinical Practice guidelines and applicable regulatory requirements. All participants provided written informed consent before participation. Given the vulnerability of participants, the study was implemented in the context of comprehensive standard-of-care treatment (described below).
Study Population
The study enrolled adults (19–64 years of age) who had a diagnosis of major depressive disorder without psychotic features according to DSM-IV-TR criteria (
30) and confirmed by the Mini International Neuropsychiatric Interview (MINI). Candidates were screened shortly after presenting to an emergency department or an inpatient psychiatric unit. Eligibility criteria required that participants respond affirmatively to MINI questions B5 (“Think about suicide [killing yourself]?”) in the present and B9 (“Intend to act on thoughts of killing yourself?”) in the past 24 hours, that they be in clinical need of acute psychiatric hospitalization due to imminent risk for suicide, and that they have a score ≥22 on the Montgomery-Åsberg Depression Rating Scale (MADRS) (
31) on day 1 before dosing. Participants had to agree voluntarily to standard-of-care treatment, including hospitalization (for 5 days after randomization, unless a longer or shorter period was clinically warranted) and initiation or optimization of one or more noninvestigational antidepressants.
Several psychiatric comorbidities were exclusionary, including a current diagnosis of bipolar disorder, moderate to severe substance use disorder, intellectual disability, antisocial personality disorder, borderline personality disorder, or a current or past diagnosis of a psychotic disorder.
Study Design
This double-blind, randomized, placebo-controlled, multicenter study was conducted from June 2014 to February 2016 at 11 study sites in the United States.
The study consisted of a 24- to 48-hour screening, followed by 4 weeks of double-blind treatment (days 1 to 25) and then 8 weeks of posttreatment follow-up (days 26 to 81). In addition to standard-of-care treatment, eligible participants were randomly assigned (1:1) to receive either intranasal esketamine (84 mg) or matching placebo, administered twice weekly. Randomization was balanced using randomly permuted blocks and stratified by study center and type of standard-of-care antidepressant (i.e., monotherapy or antidepressant plus augmentation therapy). A computerized system was used for randomization; investigators were not provided with the randomization codes.
After discharge from the inpatient psychiatric unit, visits were conducted on an outpatient basis twice weekly through day 25 during double-blind treatment, then weekly through day 52, and biweekly through day 81 during posttreatment follow-up.
Study Drug and Concomitant Antidepressant Therapy
Study drug was provided in a nasal spray device containing 200 μL of solution (i.e., two sprays). Each device delivered either 14 mg of esketamine per 100 μL spray or placebo. Three devices were used for each participant, for a total of 84 mg of esketamine. To help mask treatment assignment, a bittering agent was added to the placebo formulation to simulate the taste of esketamine.
On each double-blind dosing day, participants self-administered, under the supervision of a health care provider, one spray of study drug into each nostril three times, each 5 minutes apart. The dose of esketamine (or placebo) could be reduced from 84 mg to 56 mg for participants experiencing any intolerance.
Standard-of-care antidepressant medication was initiated or optimized for all participants on day 1. Dosage titration or adjustments of standard-of-care antidepressant treatment occurred during the first 2 weeks of double-blind treatment, with dosages maintained thereafter. Participants taking a recently initiated antidepressant at screening could continue the antidepressant at the same dosage during treatment with study drug. During the follow-up phase, participants were treated with standard-of-care antidepressant treatment, managed per clinical judgment. Participants were not permitted to receive ketamine or ECT during the posttreatment follow-up period.
Efficacy Assessments
Trained and certified site raters assessed depressive symptom severity with the MADRS, including suicidal thoughts (item 10), on day 1 (before dosing and 4 hours after dosing, according to the approach of Zarate et al. [
19,
32,
33]), on day 2 (∼24 hours after the initial dose), on all subsequent visits (before dosing), and at 4 hours after dosing on day 25 during the double-blind phase, and at all visits during the posttreatment follow-up phase.
The severity of suicide risk was assessed using the clinician global judgment of suicide risk rating from the Suicide Ideation and Behavior Assessment Tool (
34). The Suicide Ideation and Behavior Assessment Tool is a novel instrument, currently undergoing validation, that includes patient-reported and clinician-rated sections (see the
data supplement that accompanies the online edition of this article). The tool was developed to measure rapid changes in suicidality and to address the potential limitations of existing scales, including their limited sensitivity in discriminating treatment differences associated with rapid change. Information about the severity of suicidal ideation and behaviors, and risk and protective factors collected in patient-reported modules, along with a semistructured interview performed by the clinician, informed the clinician global judgment of suicide risk rating. The instrument was completed at all visits during the double-blind and follow-up phases. Suicide risk was scored from 0 (“not suicidal”) to 6 (“suicidal risk requires hospitalization with suicide precautions”).
Participants also rated their severity of suicidal ideation using the 21-item Beck Scale for Suicide Ideation at all visits during the double-blind and follow-up phases, and their sense of hopelessness using the Beck Hopelessness Scale before dosing and 4 hours after dosing on day 1, before dosing on day 25, and at the end of follow-up.
Safety Assessments
Adverse events were monitored throughout the study. Vital signs were measured and the Clinician-Administered Dissociative States Scale (CADSS) (
35) was administered before dosing and at 40 minutes, 2 hours, and 4 hours after dosing on each dosing day. With preapproval from the U.S. Food and Drug Administration, the Suicide Ideation and Behavior Assessment Tool was utilized as an alternative to the Columbia–Suicide Severity Rating Scale.
Statistical Analysis
All randomized participants who received at least one dose of study medication in the double-blind phase were included in the safety analysis set. Efficacy data were analyzed in an intent-to-treat analysis set, which included all participants in the safety analysis set who had MADRS scores at baseline and at 4 hours postbaseline on day 1.
Sample size determination.
For this proof-of-concept study, a sample size of 32 participants per treatment group was required to demonstrate a treatment difference of ≥6 points in mean change in MADRS score from baseline (day 1, predose) to 4 hours postdose between the esketamine and placebo groups, based on 91% power, a two-sided significance level of 0.20, and a standard deviation of 9. Assuming that 8% of randomized participants discontinued before providing postbaseline efficacy measurements, the total number required for each treatment group was 35.
Efficacy endpoints and analyses.
The primary efficacy endpoint—change in MADRS score from baseline to 4 hours postdose on day 1—was analyzed on last-observation-carried-forward data using the analysis of covariance (ANCOVA) model with treatment group (esketamine or placebo), antidepressant treatment (monotherapy or antidepressant plus augmentation therapy), and pooled sites as factors and baseline MADRS score as a continuous covariate.
The changes from baseline over time in MADRS score were analyzed using the ANCOVA model described for the primary efficacy endpoint. To assess the sensitivity of results from the ANCOVA analysis, a mixed-effects model using repeated-measures analysis was also performed using observed case data. The model included time, treatment, antidepressant treatment, pooled sites, and time-by-treatment interaction as factors and baseline MADRS total score as a continuous covariate. An unstructured variance-covariance matrix was used.
The MADRS suicide item and the clinician global judgment of suicide risk rating were analyzed based on ranks of change using the ANCOVA model described for the primary efficacy analysis. Change from baseline in Beck Scale for Suicide Ideation score and Beck Hopelessness Scale score was also analyzed using the ANCOVA model described for the primary efficacy endpoint. No adjustments were made for multiplicity.
Post hoc summaries of the proportion of participants achieving remission of depressive symptoms (MADRS score ≤12) and the proportion of participants with resolution of suicide risk (clinician global judgment of suicide risk score of 0 or 1) were provided.
Results
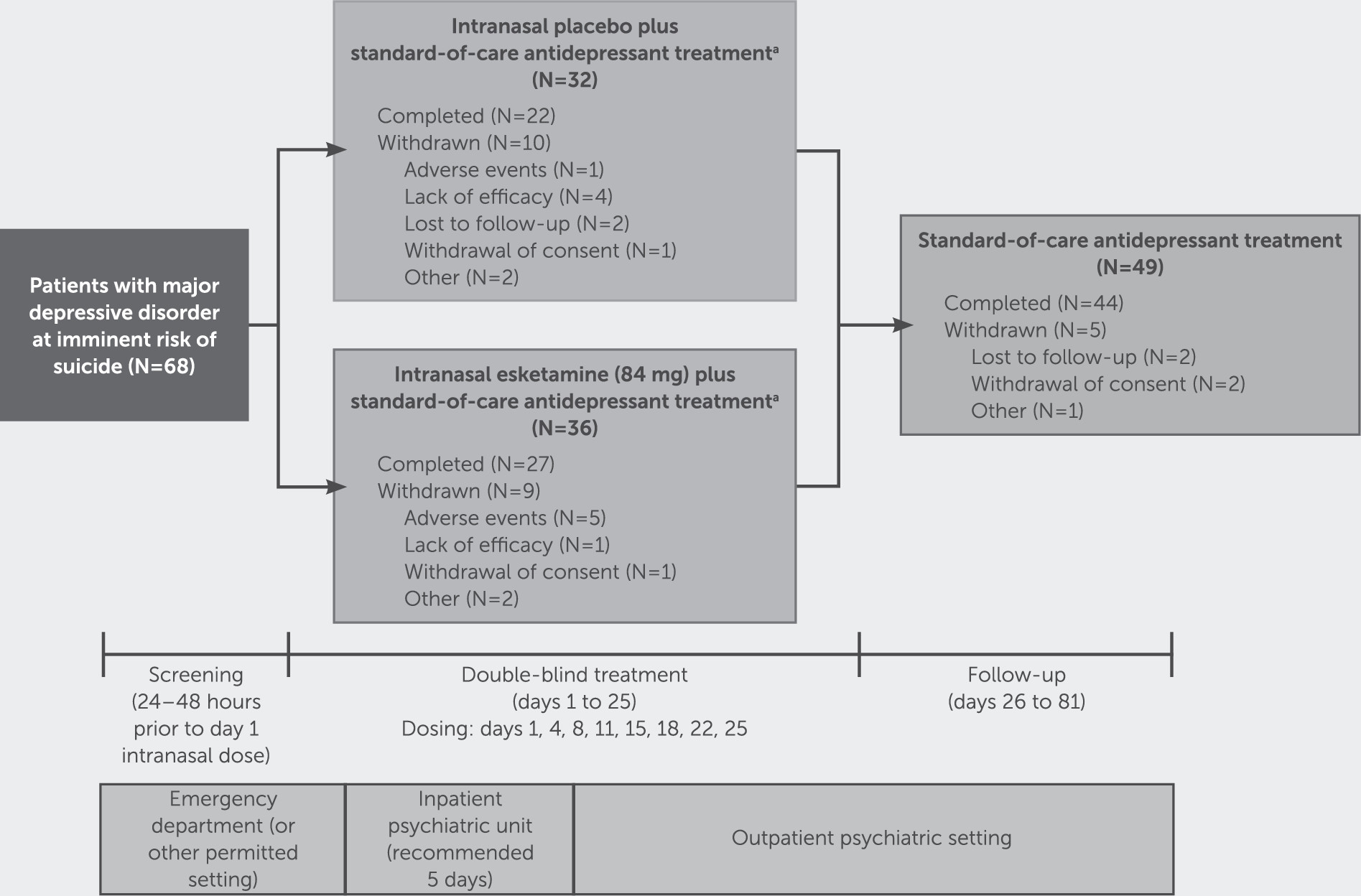
A total of 68 participants underwent randomized assignment. Of these, 66 received at least one dose of study drug (31 and 35 in the placebo and esketamine groups, respectively) (
Figure 1). These 66 participants met the criteria for inclusion in both the intent-to-treat and safety analysis sets. Most randomized participants (49/68, 72%) completed the double-blind treatment phase and entered the posttreatment follow-up phase, with 44 completing the day-81 follow-up visit.
The treatment groups were similar with respect to demographic and baseline clinical characteristics (
Table 1). The participants were severely depressed at enrollment, as evidenced by a high baseline MADRS score. All 66 participants were assessed as having current suicidal ideation with intent (“yes” responses to MINI questions B5 and B9); 44 (66.7%) reported having “thoughts about suicide (killing yourself)” that were “severe,” and 36 (54.5%) reported having such thoughts “very often.” Half of participants required suicide precautions in addition to hospitalization (a clinician global judgment of suicide risk score of 6: 57.1% in the esketamine group and 45.2% in the placebo group).
All participants received standard-of-care antidepressant treatment concomitantly with study drug during the double-blind phase. The most common antidepressant used was sertraline, in both treatment groups (22.9% in the esketamine group and 32.3% in the placebo group).
Efficacy Results
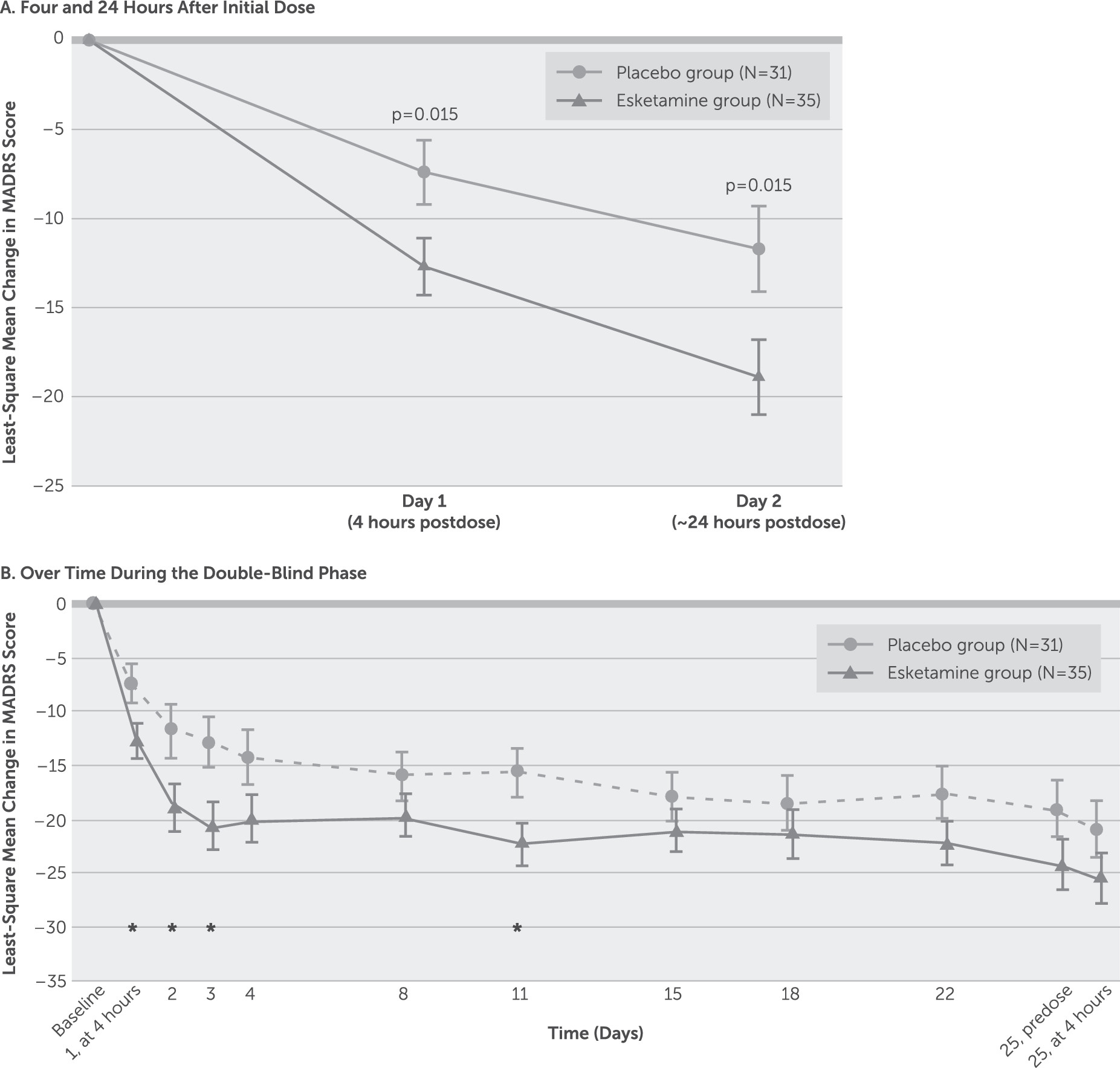
The MADRS score decreased (improved) from baseline to 4 hours after the first dose in both the esketamine group (mean=−13.4, SD=9.03) and the placebo group (mean=−9.1, SD=8.38), with significantly greater improvement in the esketamine group (least-square mean difference=−5.3, SE=2.10, p=0.015; effect size=0.61). The improvement in depressive symptoms was numerically greater in the esketamine group compared with the placebo group at all time points over the 4-week double-blind phase (
Figure 2), with significant between-group differences observed ∼24 hours after the first dose (least-square mean difference=−7.2, SE=2.85, p=0.015; effect size=0.65), at day 3 (least-square mean difference=−7.4, SE=2.92, p=0.015; effect size=0.65), and at day 11 (least-square mean difference=−7.5, SE=3.29, p=0.029; effect size=0.65), but not at the double-blind endpoint (day 25) (least-square mean difference=−4.5, SE=3.14, p=0.159; effect size=0.35). At the posttreatment follow-up endpoint (day 81), improvement in MADRS score was not statistically greater in the esketamine group compared with the placebo group (least-square mean difference, −26.3 [SE=12.74] compared with −22.4 [SE=9.44]; p=0.211). Results of the mixed-effects model using repeated-measures analysis of the MADRS score were consistent with those of the ANCOVA last-observation-carried-forward analysis.
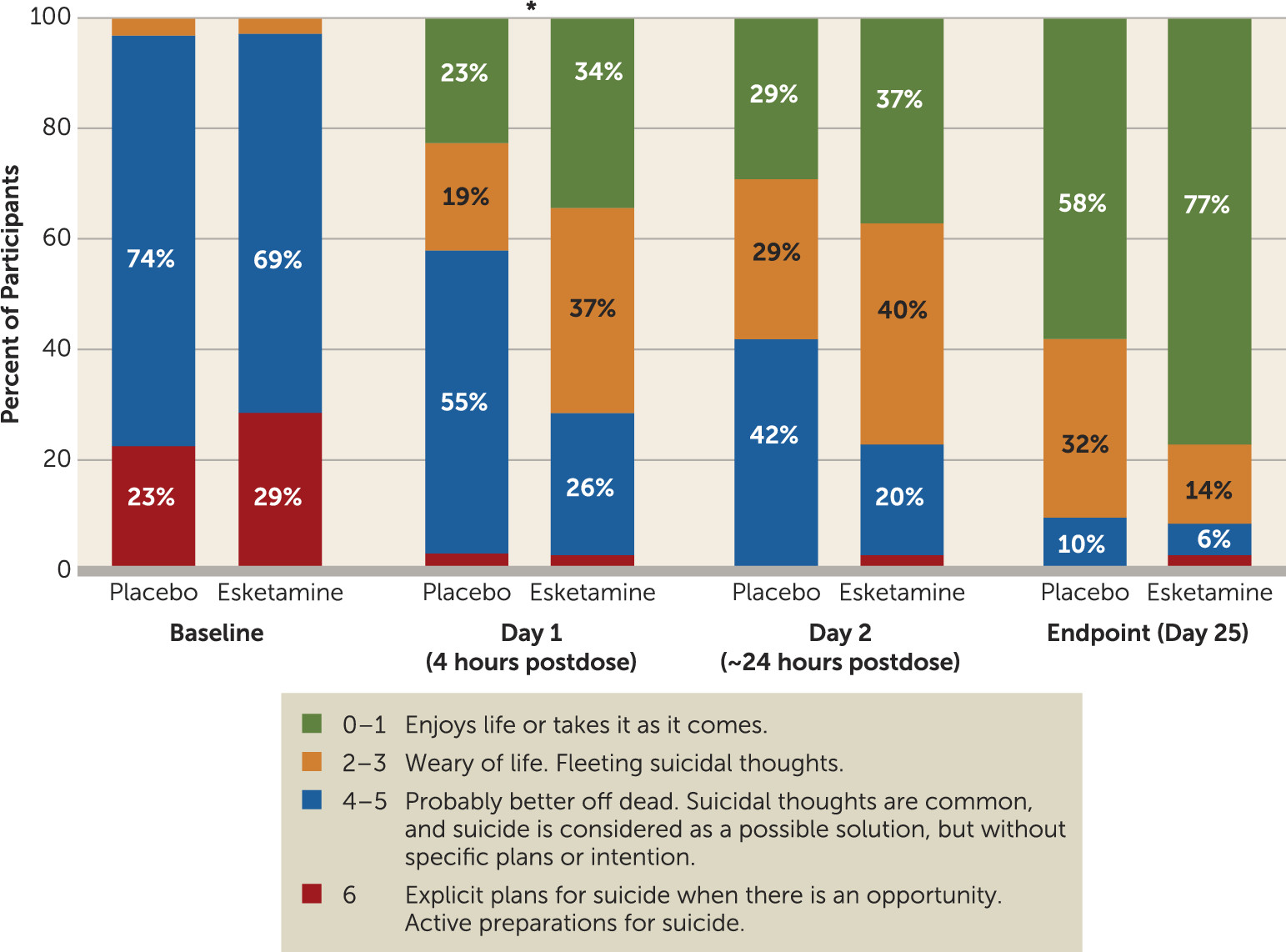
Participants in the esketamine group had significantly greater improvement in score on the MADRS suicidal thoughts item compared with those in the placebo group 4 hours after first dose (p=0.002; effect size=0.67) (
Figure 3), but not 24 hours after the first dose (p=0.129; effect size=0.35) or at double-blind endpoint (day 25) (p=0.143; effect size=0.29).
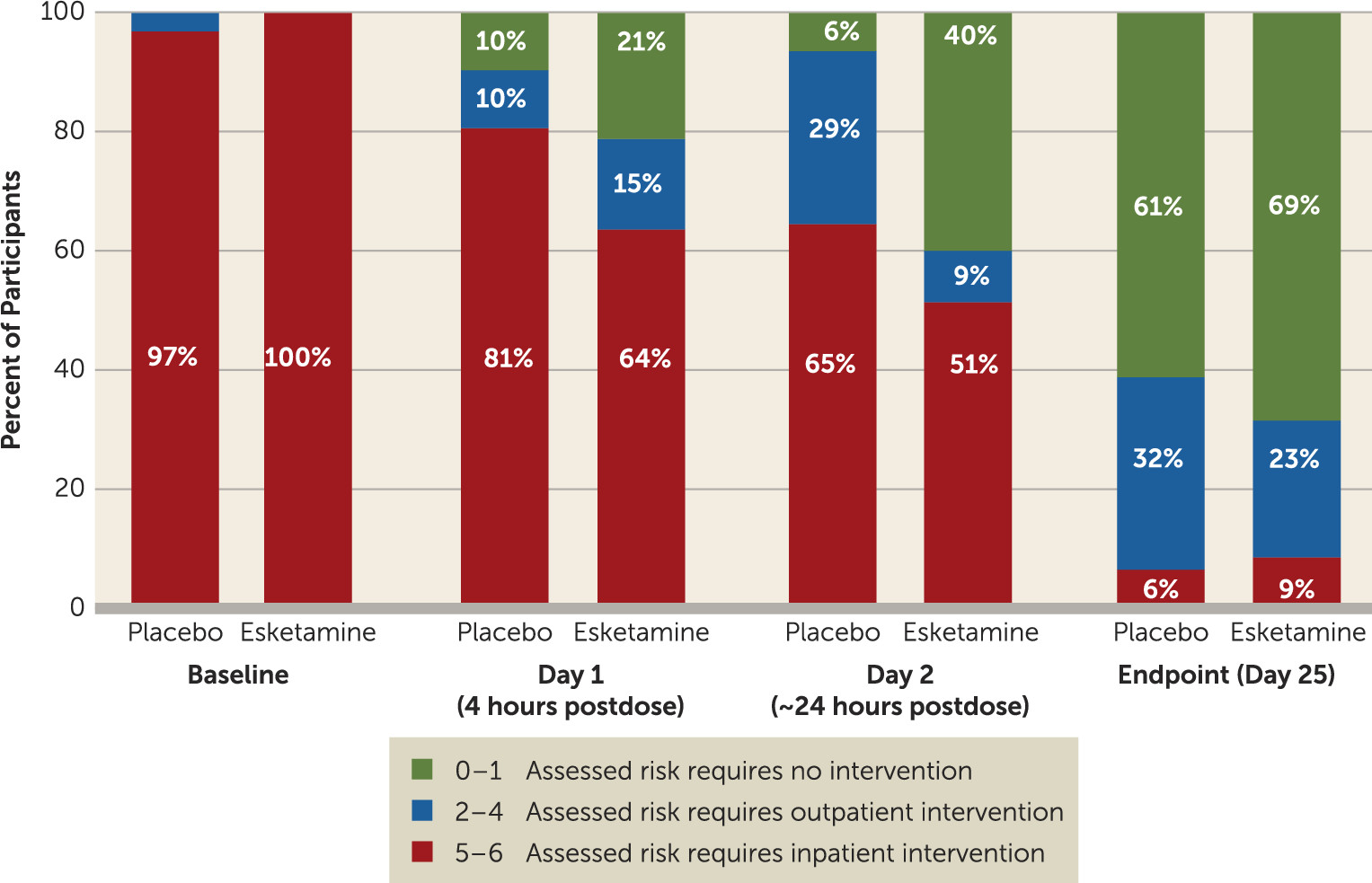
Analysis of the clinician global judgment of suicide risk rating using a ranked analysis of change from baseline showed numerically greater, but not statistically significant, decreases in ratings in the esketamine group compared with the placebo group at 4 hours (p=0.112; effect size=0.31) and 24 hours after the first dose (p=0.150; effect size=0.56). There were no differences between treatment groups at the end of double-blind treatment (p=0.922) or of posttreatment follow-up (p=0.271).
In a post hoc analysis, a greater number of participants in the esketamine group compared with the placebo group achieved resolution of suicide risk (clinician global judgment of suicide risk scores of 0–1) at 4 hours (21.2% and 9.7%, respectively) and 24 hours after the first dose (40.0% and 6.5%, respectively) (
Figure 4).
Findings from analyses of other efficacy endpoints are summarized in
Table 2.
Safety Results
Intranasal esketamine was generally well tolerated. The most frequently reported adverse events are listed in
Table 3. Adverse events leading to early termination in the double-blind phase occurred in five participants in the esketamine group (agitation, aggression, unpleasant taste, and ventricular extrasystoles in one participant each, and dizziness, dyspnea, and nausea in one participant) and one participant in the placebo group (dissociative disorder and panic attack). Three participants in the esketamine group had a dose reduction due to intolerance.
During the double-blind phase, none of the participants in the placebo group and four participants in the esketamine group experienced a serious adverse event (two had suicidal ideation, considered by investigators as doubtfully related or unrelated to study drug; one had agitation, considered unrelated to study drug; one had exacerbation of depressive symptoms, considered possibly related to study drug). During follow-up, serious events were reported for one participant in the esketamine group (suicidal ideation) and five participants in the placebo group (three made suicide attempts, one had suicidal ideation, and one had cellulitis).
Transient elevations in blood pressure were observed in the esketamine group across all dosing days (maximum mean increase from predose measurement: systolic, 16.7 mmHg [SD=10.46] compared with 8.7 mmHg [SD=7.48] for the placebo group; diastolic, 11.9 mmHg [SD=8.88] compared with 7.6 mmHg [SD=9.35] for the placebo group). Transient elevations from predose measurement in blood pressure on each dosing day peaked at 40 minutes after dosing, and typically returned to pretreatment levels within 2 hours after dosing.
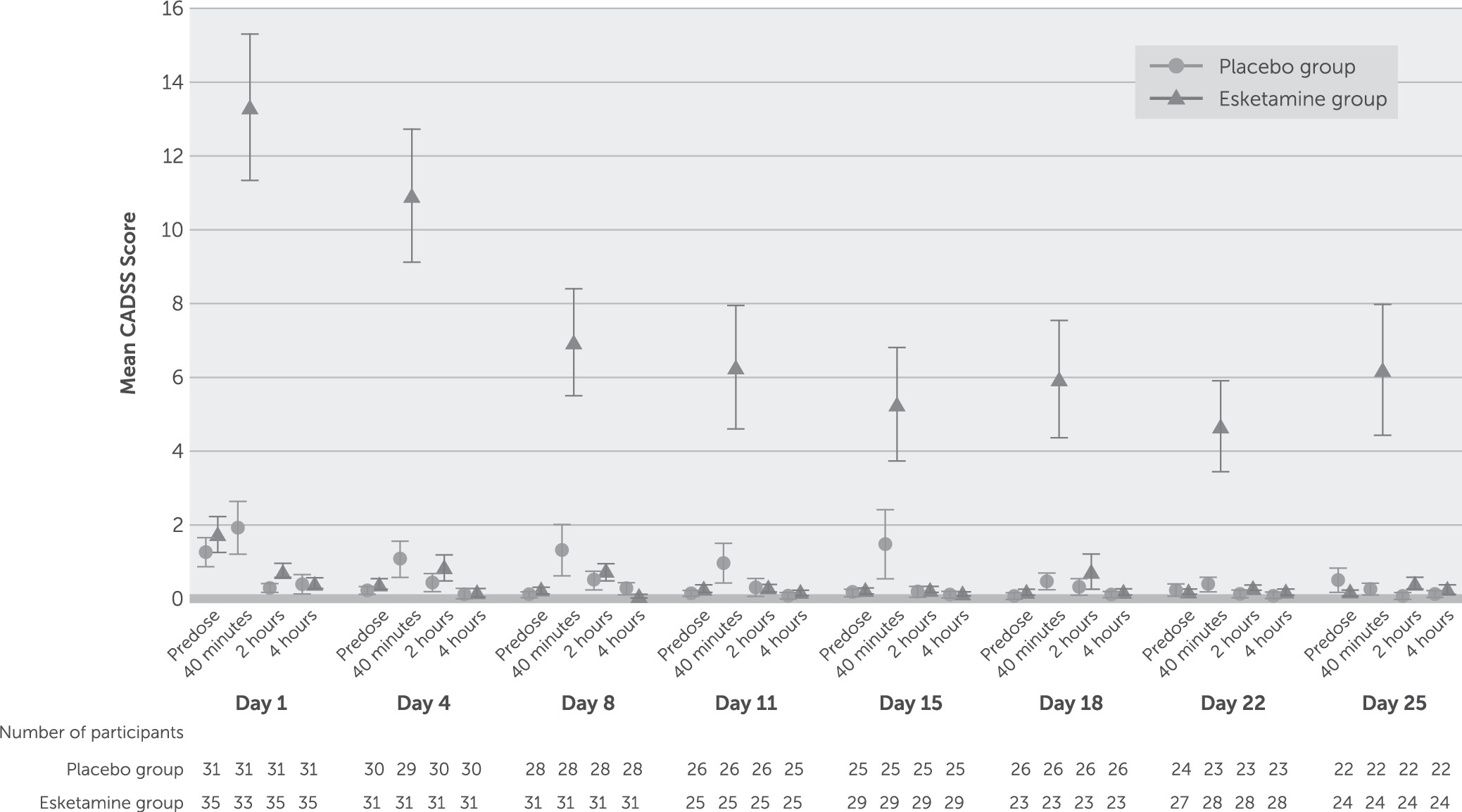
Dissociative symptoms, as measured by the CADSS, began shortly after the start of dosing, peaked at 40 minutes after dosing, resolved by 2 hours, and attenuated with repeated dosing (
Figure 5).
Discussion
The results of this proof-of-concept study support the hypothesis that intranasal esketamine may be an efficacious treatment for rapid reduction of depressive symptoms, including suicidal ideation, in patients assessed to be at imminent risk for suicide. These findings may reflect a promising breakthrough in the clinical management of a potentially lethal condition for which there are no approved pharmacotherapies.
The participants in this study, unlike those typically included in standard antidepressant trials, were severely depressed and suicidal. All enrolled individuals received comprehensive treatment, which included clinically warranted initial hospitalization and optimized standard-of-care antidepressant medication. Despite the relatively high level of clinically meaningful, nonspecific improvement observed among placebo recipients, additional beneficial effects of esketamine plus standard-of-care treatment on depressive symptoms, as measured by MADRS total score and MADRS suicide item score, were distinguished at 4 hours after the first dose. The continued numerical advantage of esketamine on these endpoints suggests that esketamine may be an important treatment to bridge the efficacy gap created by the delayed onset of action of standard antidepressants.
The clinician global judgment of suicide risk rating, a component of the Suicide Ideation and Behavior Assessment Tool, was developed to measure rapid change in suicidality, based on concerns that existing scales, such as the patient-rated Beck Scale for Suicide Ideation (or the clinician-rated version, the Scale for Suicidal Ideation), were not designed to do so. Moreover, the Suicide Ideation and Behavior Assessment Tool takes into consideration both the patient’s and the clinician’s perspectives. Similar to the Beck Scale for Suicide Ideation results observed in our study, previous trials of ketamine in depressed and/or suicidal patients found that the Beck Scale for Suicide Ideation or the Scale for Suicidal Ideation did not consistently distinguish active treatment from the comparator agent (
24,
25,
36). Specifically, a study investigating the effect of single-dose ketamine versus midazolam (as psychoactive placebo) on suicidal ideation in 24 suicidal patients with mood and anxiety spectrum disorders (
36) failed to meet its primary outcome measure, change from baseline in the self-rated Beck Scale for Suicide Ideation at 24 hours posttreatment. However, a more recent study in depressed patients with clinically meaningful suicidal ideation found a significantly greater reduction in the clinician-rated Scale for Suicidal Ideation score at 4 and 24 hours in participants receiving intravenous ketamine compared with those receiving midazolam (
26). The inconsistency of treatment effect on suicidal ideation observed across these studies may indeed be related to whether the instrument was patient or clinician rated.
Of particular interest in our study was the nearly 35% between-group difference, favoring esketamine, in the proportion of participants achieving resolution of suicide risk (i.e., a clinician global judgment of suicide risk score of 0–1) 24 hours after the first dose. This finding is consistent with results of a recently published meta-analysis of intravenous ketamine, which reported similar differences in ketamine compared with control agent in the proportion of patients without suicidal ideation 1 day after dosing (
25).
During double-blind treatment, two serious adverse events of suicidal ideation occurred in esketamine participants, and none in the placebo group. However, neither suicidal ideation event was considered by the investigator to be related to study drug, and both individuals completed the double-blind and follow-up phases with improvement in measures of depression and suicidality. Although the abuse liability of esketamine was not specifically probed for within this short-term trial, there were no reports of esketamine dependence, misuse, diversion, or withdrawal symptoms in this trial (or any of the completed or ongoing clinical trials of esketamine). However, given compelling animal data indicating the abuse liability of ketamine and esketamine (
37,
38) and the known human abuse potential of ketamine (
39), further evidence from an ongoing human abuse liability study is needed to evaluate esketamine’s abuse potential.
Within this trial, intranasal administration of 84 mg of esketamine appeared safe, with no new or unexpected safety concerns noted as compared with previously conducted treatment trials of patients with treatment-resistant depression (
28,
29). Transient elevations in blood pressure were expected based on these earlier reports. The onset of dissociative symptoms occurred shortly after the initiation of esketamine dosing, resolved by 2 hours after administration, and attenuated with repeated administration.
The study was designed as a phase 2 proof-of-concept study and thus is limited by the relatively small sample size and in its ability to truly inform clinical practice. Additionally, while the clinician global judgment of suicide risk rating from the Suicide Ideation and Behavior Assessment Tool is not yet a fully validated endpoint, a comprehensive psychometric validation program is under way. Further study limitations include enrollment of participants only in the United States and the exclusion of individuals with a history of psychotic symptoms or a current diagnosis of bipolar disorder.
Some recent studies of intravenous ketamine (
18,
26,
36) in depressed or suicidal patients have attempted to blind treatment assignment by using a psychoactive placebo. This study did not, as this approach is not entirely effective. The characteristic dissociative side effects of esketamine are difficult to mask even when a sedating benzodiazepine is used as a psychoactive placebo. As a result, some degree of functional unblinding may have biased study outcomes. Notwithstanding these limitations, this proof-of-concept study met its primary endpoint and reveals important new information regarding rapid reduction of depressive symptoms and potential improvement in suicidality with intranasal esketamine in patients at imminent risk for suicide. However, the study results should not be considered evidence of clinical effectiveness in the general population. Further investigation is needed to determine the rapid effect of esketamine on measures of suicidal ideation as well as the benefit of repeated esketamine dosing on symptoms of depression in this acutely ill patient population. Fully powered, global pivotal studies are under way of intranasal esketamine for rapid reduction of depressive symptoms, including suicidal ideation, in patients assessed to be at imminent risk for suicide.