Combinatorial Pharmacogenomic Testing Improves Outcomes for Older Adults With Depression
Abstract
Objective:
Design:
Setting:
Participants:
Intervention:
Outcomes:
Results:
Conclusions:
Objective
Methods
Combinatorial pharmacogenomic testing.
Study description.
Participants.
Statistical Analysis.
Results
Cohort description.
| Treatment Arm | |||
|---|---|---|---|
| Characteristic | TAU (N = 108) | Guided-Care (N = 98) | Total (N = 206) |
| Age (years) | |||
| Mean (SD) | 69.1 (4.3) | 69.8 (4.5) | 69.4 (4.4) |
| Median | 68.0 | 68.0 | 68.0 |
| Min, Max | 65, 85 | 65, 90 | 65, 90 |
| Sex, n (%) | |||
| Female | 79 (73.1) | 71 (72.4) | 150 (72.8) |
| Male | 29 (26.9) | 27 (27.6) | 56 (27.2) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 4 (3.7) | 4 (4.1) | 8 (3.9) |
| Not Hispanic or Latino | 104 (96.3) | 94 (95.9) | 198 (96.1) |
| Race, n (%) | |||
| White | 99 (91.7) | 89 (90.8) | 188 (91.3) |
| Black | 8 (7.4) | 8 (8.2) | 16 (7.8) |
| Asian | 0 | 1 (1.0) | 1 (0.5) |
| Other or Multiple | 1 (0.9) | 0 1 | (0.5) |
| Depression category, n (%) | |||
| None (HAM-D17 0-7) | 0 | 1 (1.0) | 1 (0.5) |
| Mild (HAM-D17 8-13) | 7 (6.5) | 11 (11.2) | 18 (8.7) |
| Moderate (HAM-D1714-18) | 33 (30.6) | 33 (33.7) | 66 (32.0) |
| Severe (HAM-D17 19-22) | 37 (34.3) | 23 (23.5) | 60 (29.1) |
| Very Severe (HAM-D17 ≥ 23) | 31 (28.7) | 30 (30.6) | 61 (29.6) |
| Psychiatric comorbidities, n (%) | |||
| Generalized anxiety disorder | 15 (13.9) | 19 (19.4) | 34 (16.5) |
| Panic disorders/Social phobia | 7 (6.5) | 12 (12.2) | 19 (9.2) |
| Post-traumatic stress disorder | 3 (2.8) | 5 (5.1) | 8 (3.9) |
| Comorbidities | |||
| Mean (SD) | 10.2 (6.9) | 10.0 (6.7) | 10.1 (6.8) |
| Median | 9.0 | 9.0 | 9.0 |
| Min, Max | 0.0, 45.0 | 1.0, 47.0 | 0.0, 47.0 |
| Cardiovascular comorbidities | |||
| Mean (SD) | 0.2 (0.5) | 0.2 (0.5) | 0.2 (0.5) |
| Median | 0.0 | 0.0 | 0.0 |
| Min, Max | 0.0, 3.0 | 0.0, 2.0 | 0.0, 3.0 |
| Severity of comorbidities | |||
| Mean (SD) | 0.7 (1.0) | 0.7 (1.0) | 0.70 (1.0) |
| Median | 0.0 | 0.0 | 0.0 |
| Min, Max | 0.0, 4.0 | 0.0, 4.0 | 0.0, 4.0 |
| HAM-D17 score | |||
| Mean (SD) | 20.2 (4.9) | 19.4 (5.1) | 19.8 (5.0) |
| Median | 20.0 | 19.0 | 19.5 |
| Min, Max | 8.0, 32.0 | 7.0, 29.0 | 7.0, 32.0 |
| Failed medication trials | |||
| Mean (SD) | 3.7 (3.2) | 3.3 (2.6) | 3.5 (2.9) |
| Median | 3.0 | 3.0 | 3.0 |
| Min, Max | 1.0, 23.0 | 1.0, 14.0 | 1.0, 23.0 |
Outcomes.
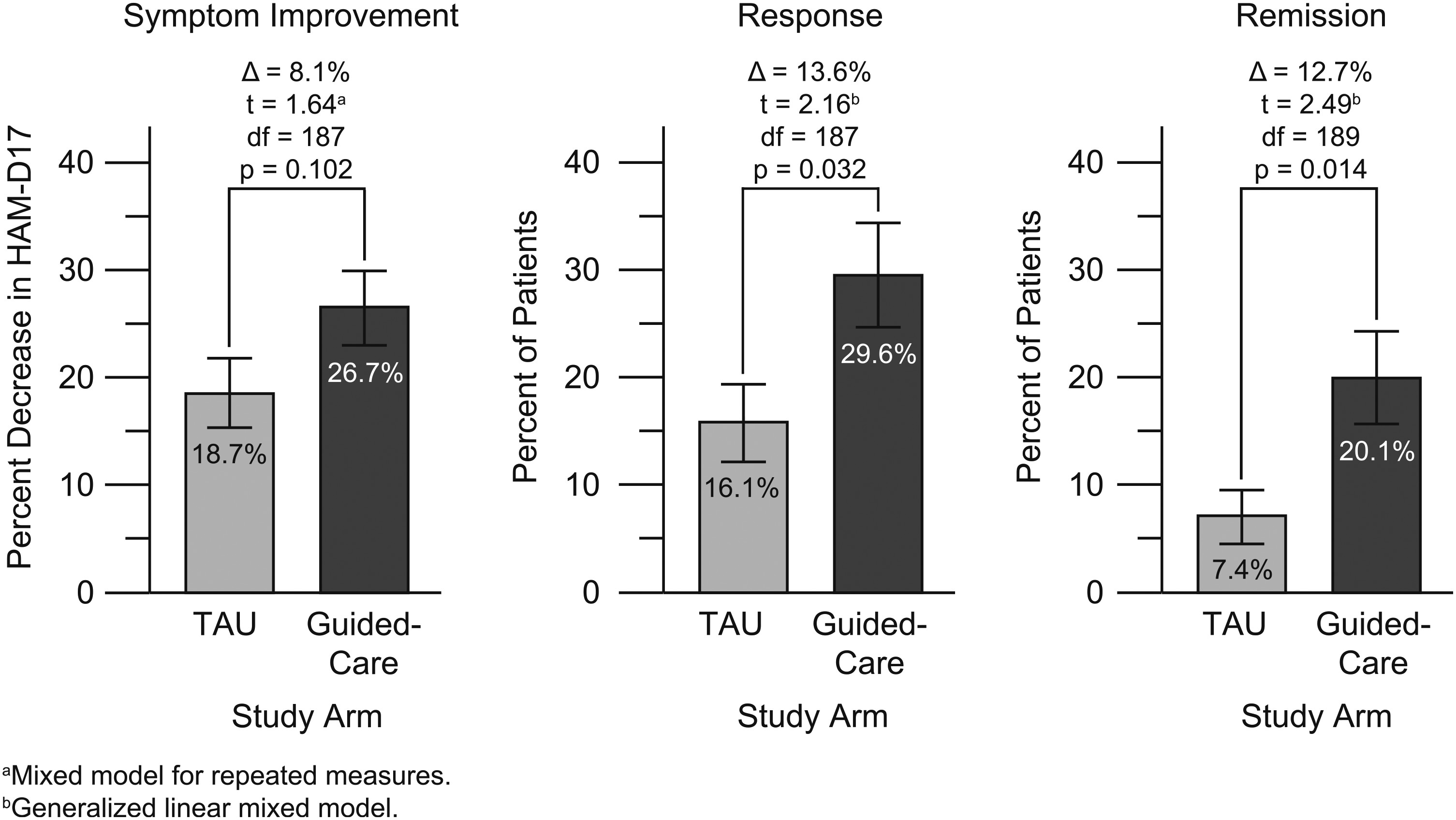
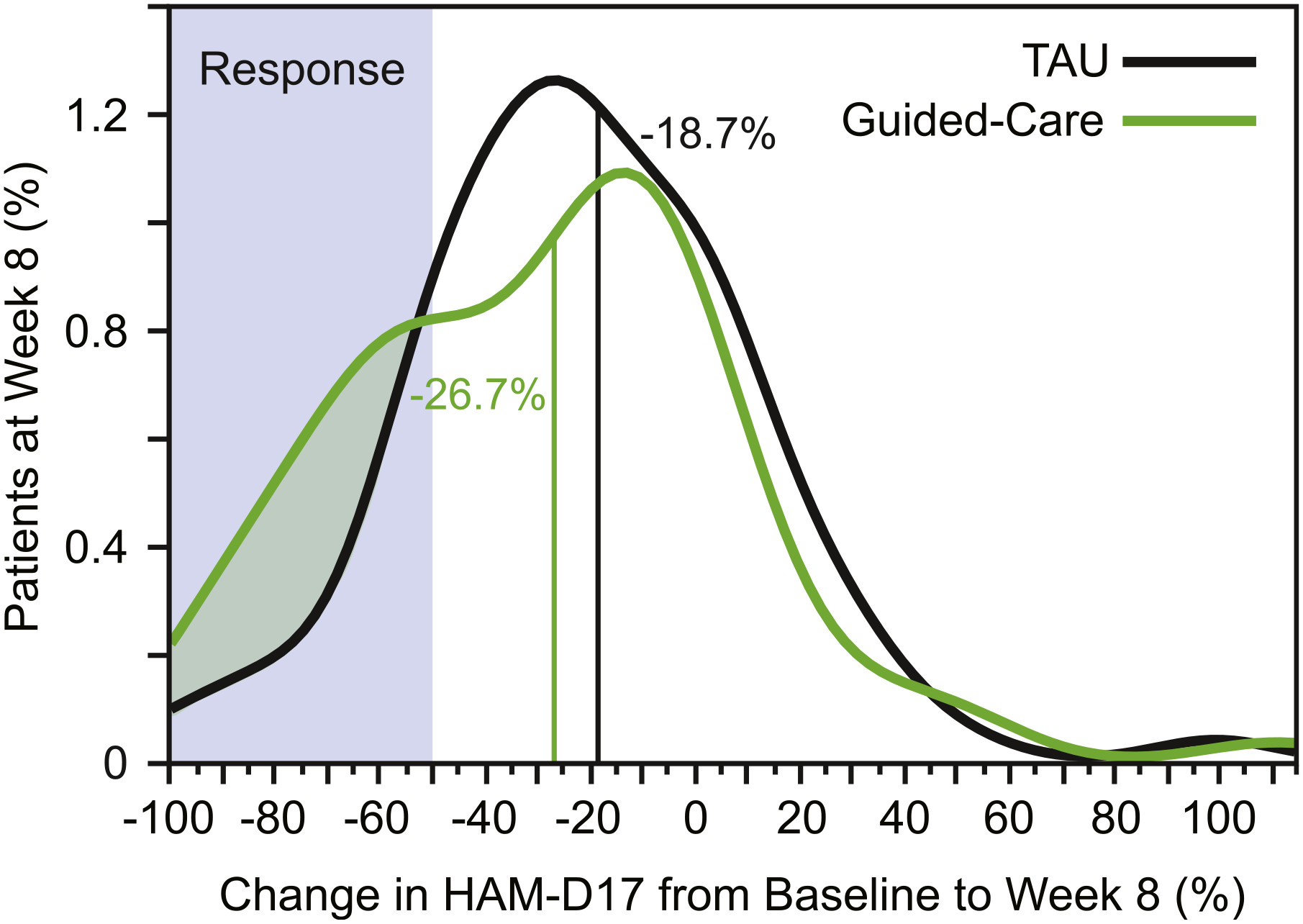
Predicted GDIs.
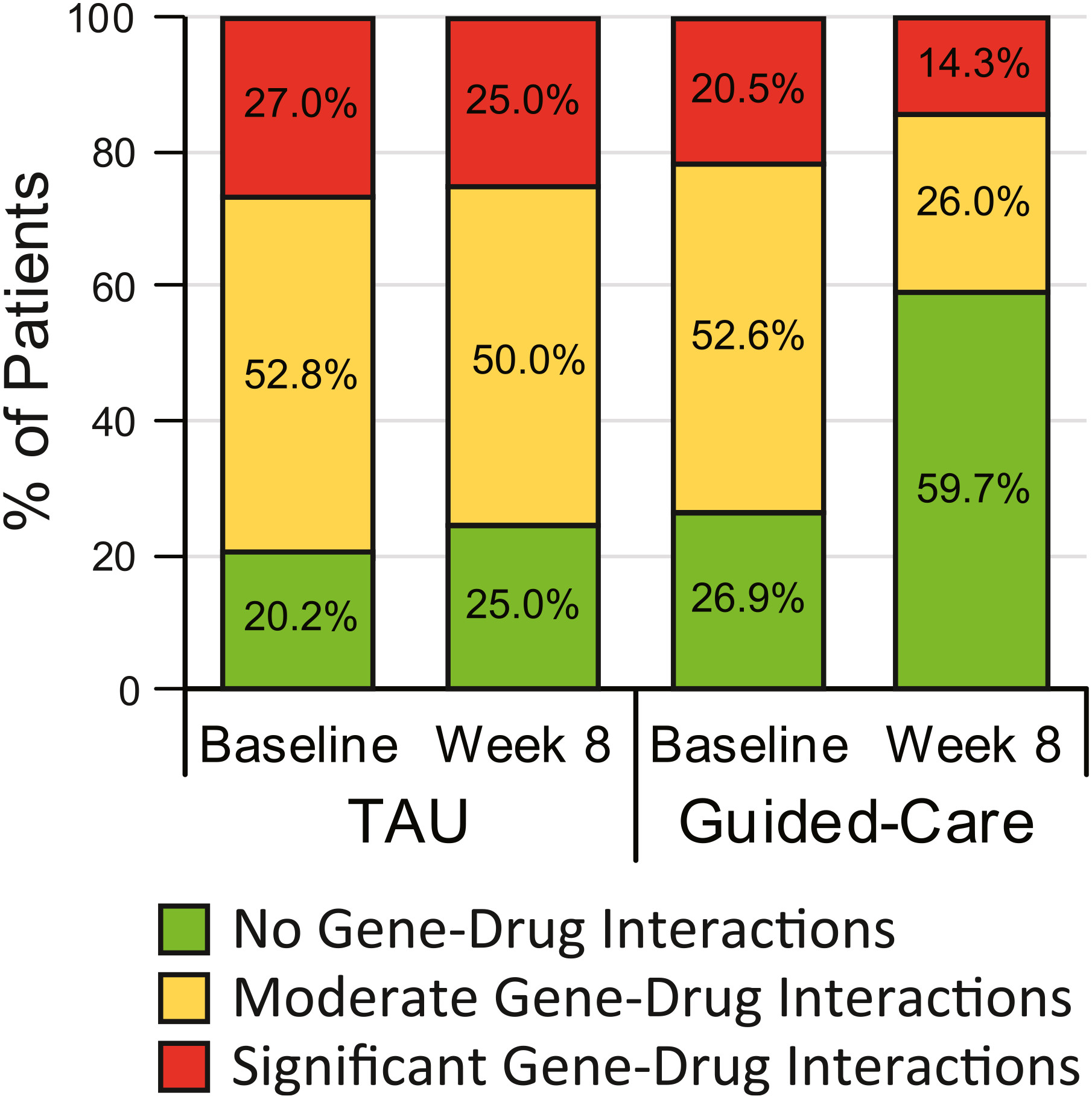
Moderation by comorbidities and concomitant medications.
| Moderator Effect | Main Effect | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Symptom Improvement (F, p-value)a | Response (F, p-value)b | Remission (F, p-value)b | Symptom Improvement (F, p-value)a | Response (F, p-value)b | Remission (F, p-value)b | df for All (Numerator, denominator) | ||||||
| Number of all comorbidities | 1.26 | 0.264 | 1.91 | 0.169 | 0.55 | 0.460 | 0.35 | 0.552 | 1.99 | 0.160 | 1.55 | 0.214 | 1, 179 |
| Number of cardiovascular comorbidities | 0.36 | 0.549 | 0.56 | 0.456 | 0.00 | 0.978 | 0.67 | 0.414 | 0.00 | 0.982 | 0.40 | 0.527 | 1, 179 |
| Severity of comorbiditiesc | 0.04 | 0.838 | 0.11 | 0.740 | 0.89 | 0.348 | 1.19 | 0.277 | 0.08 | 0.771 | 0.00 | 0.990 | 1, 179 |
| Number of concomitant medications | 0.41 | 0.524 | 0.69 | 0.406 | 0.20 | 0.654 | 0.32 | 0.571 | 0.30 | 0.585 | 0.01 | 0.940 | 1, 178 |
Durability of combinatorial pharmacogenomics-guided care utility.
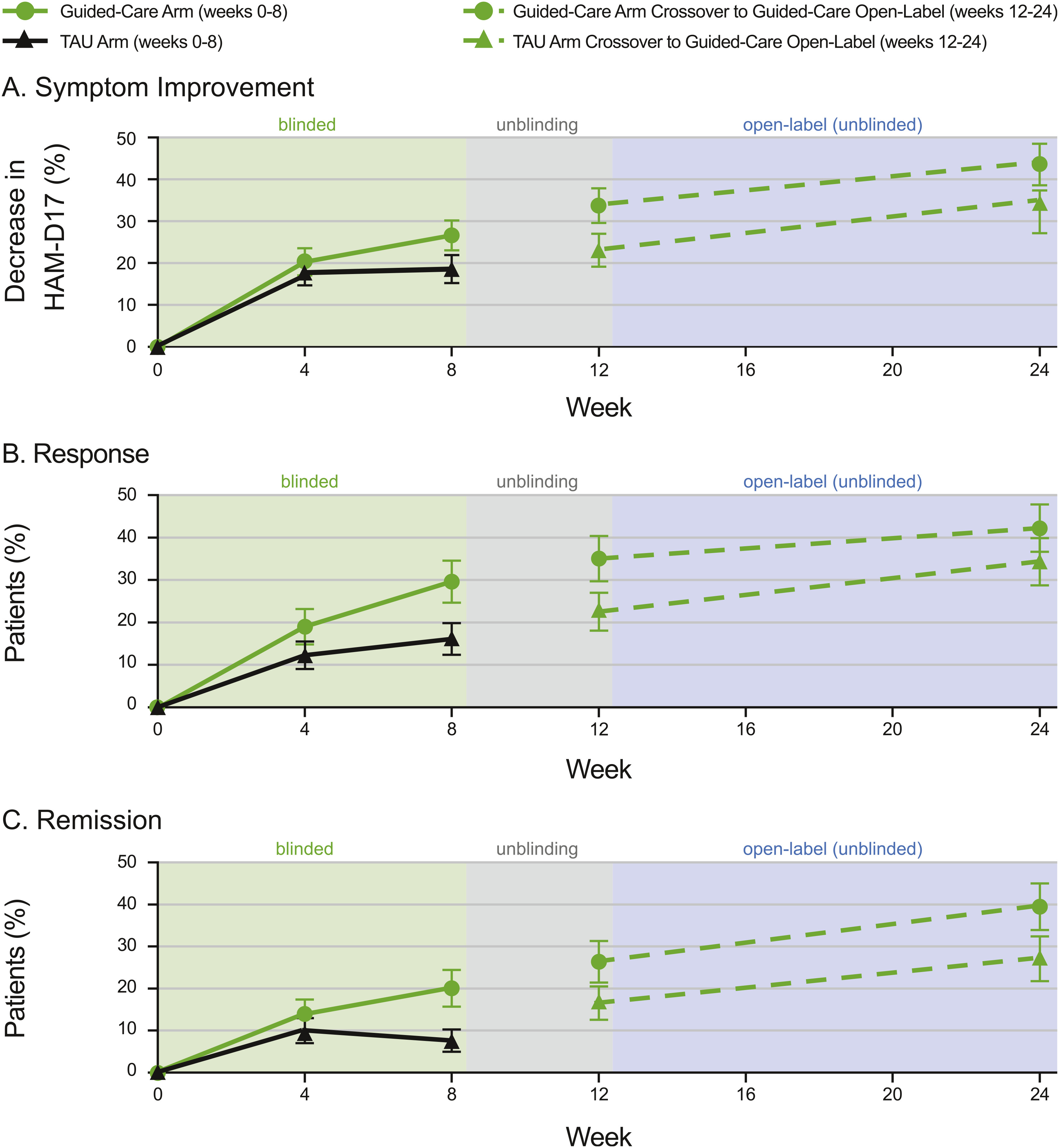
Conclusions
Disclosure
References
Information & Authors
Information
Published In


History
Keyword
Authors
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBGet Access
Login options
Already a subscriber? Access your subscription through your login credentials or your institution for full access to this article.
Personal login Institutional Login Open Athens loginNot a subscriber?
PsychiatryOnline subscription options offer access to the DSM-5-TR® library, books, journals, CME, and patient resources. This all-in-one virtual library provides psychiatrists and mental health professionals with key resources for diagnosis, treatment, research, and professional development.
Need more help? PsychiatryOnline Customer Service may be reached by emailing [email protected] or by calling 800-368-5777 (in the U.S.) or 703-907-7322 (outside the U.S.).