Alzheimer’s disease (AD) has always been a primarily clinical disease, seeing as its confirmation could only be reached through histopathological post-mortem studies. However, the more its physiopathology is known, the more certainty should be put into diagnosing it. Therefore, scientists have searched for biomarkers to help as diagnostic tools. Nevertheless, the fast rise of biomarkers gave rise to many questions such as, what biomarkers exist today? How can they be used in AD’s diagnosis? When does AD really start? In this review, we aim to answers those questions.
The Inclusion of Biomarkers in the Diagnostic Criteria
As the knowledge on the pathophysiological, molecular and structural changes in AD increased, Dubois et al. (
8) revised AD’s classical diagnostic criteria and proposed new ones. These new diagnostic criteria aimed to identify AD in its earlier stages, before the development of a dementia syndrome. They established a specific clinical phenotype of AD, casting aside the diagnosis of exclusion and solving the problem of low diagnostic specificity, while also offering the chance of an early therapeutic intervention (
8). Their principal criterion is a failure in episodic memory that appears early in the disease (A, see
Table 1). Furthermore, they introduced as a novelty the support criteria which are based on biomarkers (B, C, D, and E). Therefore, the presence of the main criterion together with at least one of the supporting criteria is indicative of AD pathology.
With these criteria, Dubois et al. (
8) arrive at the diagnosis of AD earlier and in a more specific way than their predecessor, the NINCDS-ADRDA criteria, covering the earliest stages of the disease, when there is still no dementia syndrome, as well as the later stages when the patient is already functionally disabled. The ingenuity resides on the fact that biological biomarkers are included in the criteria for the first time, as a requirement to reach diagnosis. These biomarkers include structural and molecular imaging, cerebrospinal fluid (CSF) analysis, and genetic mutation analysis.
After Dubois’ first step in including biomarkers as diagnostic criteria, in 2011, the National Institute on Aging and the Alzheimer’s Association (NIA-AA) defined new diagnostic criteria that separated the disease in three clinical stages, each one with its own diagnostic recommendation. First is the preclinical stage that presents pathologic brain changes, which may be in progress decades prior to disease, without evident clinical symptoms. In this stage, alterations can be seen in CSF and imaging biomarkers although, at present, they cannot predict which of these individuals will develop dementia (
9). The second stage is Mild Cognitive Impairment (MCI), which is marked by memory symptoms that are greater than normal for a person’s age and education but that do not interfere with their independence and may or may not progress to Alzheimer’s dementia (
10). The final stage is Alzheimer’s dementia, in which symptoms are significant enough to impair a person’s ability to function independently (
11).
The definition of a preclinical stage that could be in progress many years before the beginning of symptoms meant that the biomarkers acquired a greater importance in the diagnostic process.
When Does AD Really Start?
With the advent of biomarkers came a conceptual change of the disease. We moved from a “static and defensive” view of the pathogenesis of AD to a “dynamic and compensatory” point of view. In the first viewpoint, the brain lesions that lead to neuronal and synaptic loss and finally to cognitive deterioration depend on the degree of external aggression and on the structural reserve that each person has. The current view considers an inter-individual variability in the response to these initial aggressions, as well as differences in the severity of the pathological process and in the efficiency and evolution over time of the cerebral compensatory mechanisms (
83–
85). Therefore, the idea that AD’s main pathological processes have already taken place before we can clinically diagnose MCI has been established, and this is reinforced by the fact that these lesions begin even decades before the appearance of the earliest symptoms, when the subject is still cognitively normal (
86). Therefore, this change in perspective increasingly supports the need for early therapeutic action in order to compensate for those biological processes that are already compromised before the onset of the cognitive failure (
87).
AD is now considered a neurodegenerative disease with a very long evolution that starts silently decades before the onset of symptoms and advances gradually and slowly until it compromises the person’s cognition. Therefore, we moved from a static vision of AD in which a person is affected or not by the disease, to a dynamic concept of AD, in which dementia is considered the final stage of a set of pathological changes that occur in a chronic and gradual manner.
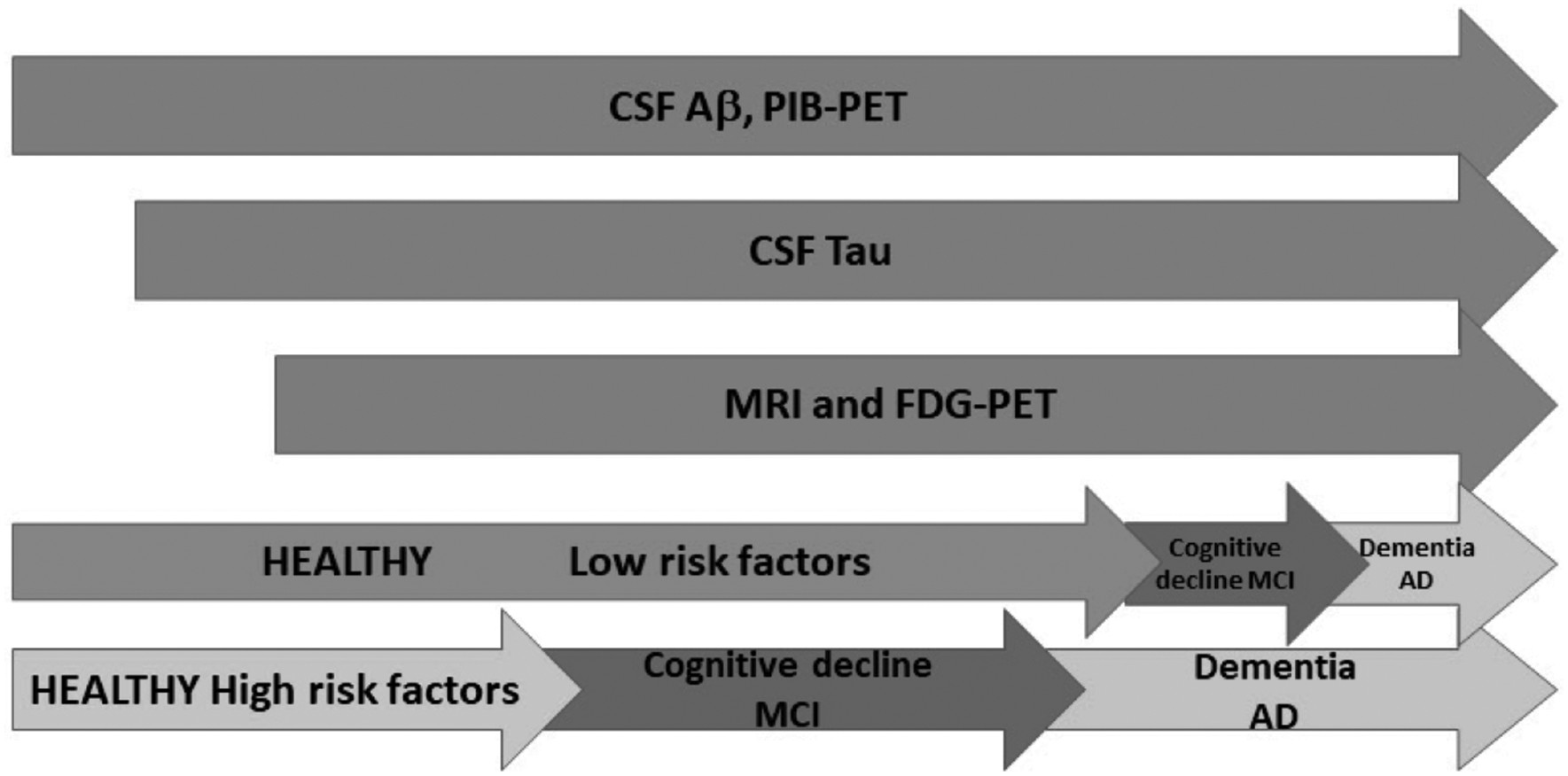
In this progression, which may take years, biomarkers can anticipate the clinical manifestations of dementia and, as the new diagnostic criteria introduced biomarkers in a supporting role in AD’s diagnosis, many laboratories worldwide have already started using them. With this in mind, and based on determinations made in different populations, Jack and collaborators proposed a model for the evolution of AD over time, known as “the dynamic biomarker cascade model” (
16). In this model, biomarkers do not increase all at once but do so in an orderly manner, a concept that is reinforced in the work by Dubois and collaborators (
18). The model presents three phases along the continuum of the disease—first, the cognitively normal asymptomatic phase, then the MCI phase that begins to show clinical affectation, and finally the dementia phase. All over this spectrum, the biomarkers described previously would present abnormal levels as the disease evolves and, eventually, correlate with the clinical symptoms presented by the patients.
Jack and collaborators propose as the initial event the abnormally increased levels of Aβ that would lead to the formation of cerebral amyloid plaques. This would be reflected in the decreased levels of Aβ in CSF and in the increased amyloid load in PiB-PET, and these alterations would appear while the individuals are still cognitively normal. Afterward, there would be an increase in CSF tau abnormalities followed by alterations in FDG-PET. These are biomarkers of neuronal dysfunction and neurodegeneration and correlate with the severity of clinical symptoms. Lastly, in advanced stages, structural brain changes would appear such as cortical atrophy and decreased hippocampal volume that could be detected by MRI (
16).
A very recent work by Petrella and collaborators (
88) developed a mathematical causal model of the dynamic biomarker cascade theory in AD, which might help to explain how these biomarkers interact and evolve over time and could potentially help patients, researchers, and medical personnel. This is a great advancement in the knowledge of the disease, but there is still a long way to go. Although, biomarkers could have a role in predicting whether a patient could convert from MCI to AD, there is not a consensus on which biomarkers could assume that role (
89,
90).
However, the scientific community’s efforts go beyond designing computational models to determine the behaviour of different biomarkers in the evolution of the disease. Models have been designed for many different aspects of the disease, such as a model based on the amyloid cascade hypothesis, showing the effects of pathological processes such as oxidative stress, inflammation or cerebrovascular disease in the kinetic of Aβ aggregation (
91). Moreover, another model focused on synaptic loss and compensation by the reinforcement of the remaining connections (
92) and, more recently, Ding et al. (2018) designed a hybrid computational approach for a more accurate disease severity classification (
93).
Nevertheless, as scientists started to better understand AD’s pathophysiology, the biggest challenge became designing computational models capable of predicting the efficacy of a specific treatment. To reach this objective, models have been created analysing potential treatments. Anastasio (2013) incorporated the role of estrogens in Aβ regulation into a model that can generate therapeutic predictions and the possible benefits of this therapy (
94). This model showed that estrogen could reduce Aβ and that non-steroidal anti-inflammatory drugs could provide a small additional benefit.
Furthermore, immunotherapy, probably the most promising treatment for AD at this moment, was also analysed by computational models. Diem et al. (2016), have incorporated this therapy’s possible complications into their model and concluded that a failure in periarterial drainage seems to be an important mechanism (
95). Another computer simulation model pointed out that immunotherapy against Aβ might not be effective, unless it is used during early stages of AD (
96) or combined with other therapies. However, a more recent model simulated the differential impact of Aβ oligomers on glutamate and nicotinic neurotransmission while under different treatments, including a passive vaccination with the monoclonal antibody solanezumab, the use of the beta-secretase inhibitor verubecestat, and of the gamma-secretase inhibitor semagacestat. They predicted a cognitive worsening in people with low Aβ baseline and an improvement in those with moderate to high Aβ levels (
97).
Computational models analyzing neurotransmitters have also been created. One such model has been implemented using preclinical data available on receptor pharmacology of cholinergic and catecholamine neurotransmitters and clinical data, to predict the effects of memantine, an N-Methyl-D-aspartic acid (NMDA) inhibitor, in different phases of AD pathology (
98).
Finaly, Stefanovski et al. (2019) created a computational multi-scale brain model, using the Virtual Brain Platform, and including PET and electroencephalogram, to simulate regional neural activity and hyperexcitability in AD and how it relates to Aβ. This model reveals a potential functional reversibility of large-scale alterations in AD after memantine treatment (
99).
Abnormalities in Biomarkers Precede Clinical Symptoms
In the “dynamic biomarker cascade model,” each biomarker reaches its maximum effect at a certain moment in the progression of the disease, and that happens in an orderly manner over time. Interestingly, the maximum levels can be detected in a person before any clinical symptom. In fact, several studies have shown that 20–40% of cognitively normal old present Aβ deposits in their cerebral tissue (
22,
100,
101). Moreover, in post-mortem samples from non-demented elderly people, Aβ plaques were also present (
102,
103). Therefore, deposits of amyloid plaques alone, even in significant quantities, are not enough to produce dementia (
16,
26,
27,
30). Not only Aβ but also tangles may be present in subjects without cognitive decline. Nonetheless, in asymptomatic patients, the presence of neurofibrillary tangles tends to be limited to the entorhinal cortex (stages of Braak I–II), while in symptomatic subjects, tangles are much more widespread (
102,
103).
On the other hand, data obtained through imaging studies with PiB-PET suggests that Aβ deposits may appear up to two decades before the onset of clinical manifestations of dementia (
86). This concurs with the fact that, after the diagnosis of AD, the levels of Aβ in CSF do not change significantly, since it has already reached a plateau, and also with the pattern of PiB retention, which is not modified throughout the disease’s spectrum (
86). Consistent with the previous studies, investigations on familiar AD have shown that these alterations in Aβ and p-tau precede by more than 10 years the clinical onset of the disease (
104). Very recently, sequential changes in normal older adults, from Aβ to tau to cognition, have been described using repeated tau-PET and amyloid-PET measures to detect the earliest AD pathologic changes (
105).
These studies are all in line with a final concept that this model postulates—the existence of a latent phase of variable duration between plaque formation and the onset of the neurodegenerative cascade. This could be due to differences in the processing of Aβ, to the capacity of resistance to pathological damage derived from the toxicity of Aβ and to compensatory mechanisms (
106).
There is an increasing idea that AD pathology would trigger cerebral compensatory mechanisms all across the AD spectrum, and it would be in the preclinical phase that these mechanisms would begin to appear. However, there is still no consensus regarding the role that compensatory mechanisms might play in cognitively healthy subjects at risk of AD with positive biomarkers, and also regarding the influence they could have on the conversion to dementia. Lazarczyk et al. (
104) suggest that the compensatory mechanisms would be divided into two categories: the passive ones (matching the cognitive reserve concept) and the active ones. The former would delay conversion to dementia, and the latter could stop disease progression in the preclinical phase and effectively prevent conversion to dementia.
Anatomically, this compensatory mechanism can be seen in structural changes found in asymptomatic people carrying the presenilin 1 mutation when compared to age-matched controls (
107). These individuals present a cortical thickening mainly in temporal and parietal areas, extending into precentral and postcentral cortex and pars triangularis, and also in structures of the posterior midline, such as precuneus and posterior cingulate. No areas of cortical thinning are observed in asymptomatic carriers, unlike those found in symptomatic people. Furthermore, structural changes are not limited to subjects with mutations in AD determinant genes, since they are also seen in the sporadic form of the disease. Some studies have shown that healthy subjects with evidence of initial deposits of Aβ (
108) present greater volume and thickness in AD related cortical regions. With AD progression, these areas suffer a progressive thinning of gray and also white matter reaching the atrophy observed in more extensive regions in the symptomatic stage (
109).
It would be interesting to have more studies evaluating these compensatory mechanisms, to understand it better and, if possible, to add it to the current computational models evaluating AD’s pathology and possible treatments, as they seem to be present since the very early stages of the disease.
New AD Classification Based on Biomarkers
Biomarkers provide a powerful tool for the clinical practice as well as for research, especially in human clinical trials, by improving diagnosis. This knowledge leads to an increase in clinical trials evaluating possible drugs that could cure, ease or control AD, but the efforts are still challenging.
Since the gap between the pathological processes and the cognitive symptoms has proven itself quite large, and the risk of generating hypotheses that were not based on the pathophysiological changes of AD was a reality, the NIA-AA (
113) proposed a new framework for the use of biomarkers in observational and interventional research. Therefore, the A/T/N system was described classifying subjects according to the number of positive biomarkers they presented. This system includes a binary system (positive or negative) depending on the measured biomarker. “A” refers to the value of a β-amyloid biomarker (amyloid PET or CSF Aβ42), “T” to the value of a tau biomarker (CSF p-tau, or tau PET), and “N” to biomarkers of neurodegeneration or neuronal injury (18FDG–PET, structural MRI, or CSF total-tau) (
114). A person with a positive “A” biomarker is classified as being in the “Alzheimer’s continuum”, that denotes either Alzheimer’s pathologic changes or AD, and those with positive biomarkers in both “A” and “T” categories are classified as having AD (
113). The possible outcomes of this classification system are illustrated in
Table 2.
A recent study analysing the prevalence of biologically defined AD compared to clinically defined probable AD reports that the former is more prevalent than the latter, especially at age 85 years, a difference that is mostly driven by asymptomatic individuals with biological Alzheimer disease, that could be diagnosed after the A/T/N description (
115).
Therefore, this research framework defines AD by its pathological alterations that could be documented by biomarkers and not by its clinical consequences. Although, at the moment, it is intended only for research and not routine clinical care (
113,
116), this new viewpoint might improve the selection of subjects and the conception of new therapies for clinical trials.