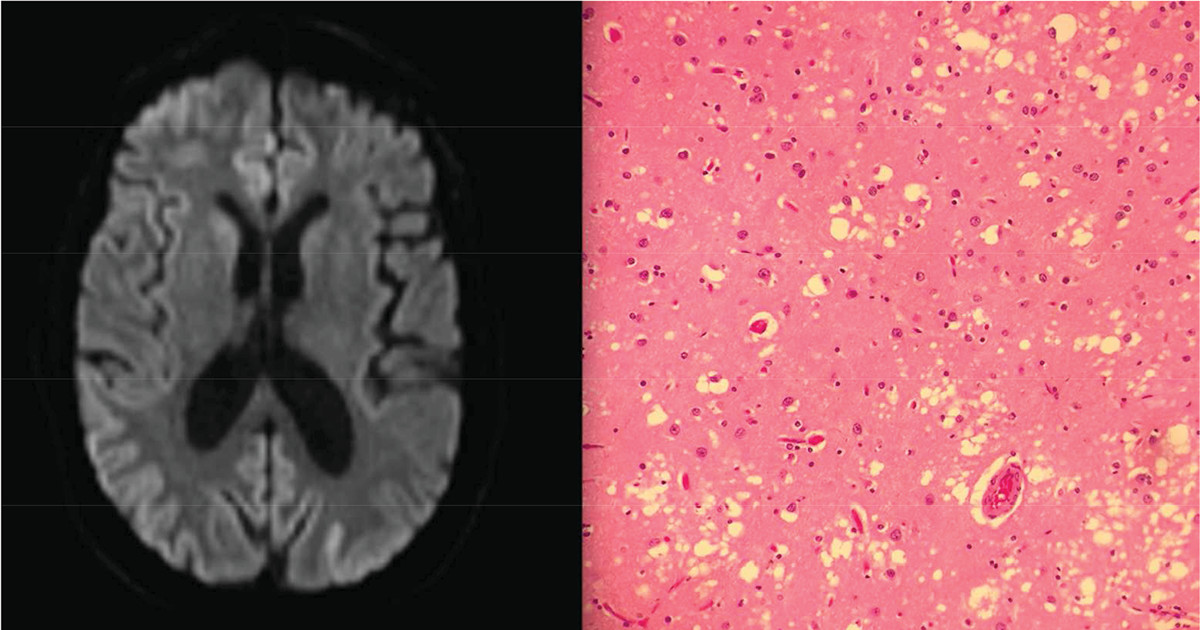
In approximately one of four cases of sCJD, the presenting symptom is psychiatric or behavioral.
2 As discussed by Ali et al.,
2 it is not uncommon for patients with sCJD to be initially misdiagnosed with a primary psychiatric disease and admitted to a psychiatric unit, as was the case with our patient. His history of poor financial and legal decisions is consistent with the executive dysfunction, poor judgment, and impulsive behavior seen in sCJD. Given the success our patient achieved in his career, his poor decision making resulted in lawsuits brought against him and subsequent disbarment.
Our patient’s prolonged course of at least 48 months is atypical for sCJD. Of those with sCJD, 85% die within 12 months of symptoms.
3 Brown et al.,
6 in their consecutive series of 230 patients with neuropathologically verified CJD, reported only five patients with duration of symptoms longer than 4 years. In this series, the median duration of symptoms was 4 months and the mean duration was 7.6 months; 90% of patients died within 1 year of symptom onset.
6 Our patient’s symptom duration is even more notable when considering his sCJD molecular subtype, MM1. Parchi et al.
4 originally published 186 cases of MM1- and MV1-type sCJD with a mean symptom duration of 3.9 months and a range of 1–18 months. MM1 and MV1 were combined, given their similar pathological features.
7 In their 2009 series, Parchi et al.
5 reported 66 cases of MM1- and MV1-type sCJD with a mean symptom duration of 4.0 months and a range of 1–24 months. Subtypes other than MM1 are associated with significantly longer duration. The longest mean symptom duration by subtype reported by Parchi et al.
5 was 20.0 months, associated with the MM/MV 2C subtype. Thus, we report the longest duration of symptoms for sCJD associated with the MM1 subtype.
The variable and oftentimes subtle first manifestations of the disease may limit the establishment of a clear onset of symptoms and thus an accurate duration of symptoms. Our patient’s profession provided opportunities for early behavioral changes to manifest themselves in legal and financial difficulties. This allowed symptom onset to be more easily established. In addition to molecular subtype, reasons for variable duration of symptoms in sCJD are unclear. One consideration in this case is that our patient’s age and educational attainment may have influenced his symptom duration via cognitive reserve, although this has not been documented in sCJD. That other genetic factors may influence symptom duration is supported by previously reported racial differences.
8,9 Furthermore, both new variant CJD, a relatively new phenotype of CJD, and familial prion diseases are commonly associated with longer symptom duration than sCJD.
7,10 This case suggests the importance of factors other than molecular subtype, whether environmental, demographic, or other genetic modifiers, in determining sCJD symptom duration.