Turner syndrome, the most common sex chromosome aneuploidy (1:2000 female live births), is caused by 45X monosomy (60%), 45X/46XX mosaicism (30%), or other X chromosome abnormalities (10%). Most affected females have short stature and impaired ovarian function, often presenting with primary amenorrhea and underdeveloped secondary sex characteristics at the time of puberty. Distinctive features include webbed neck, broad chest, cubitus valgus, widely spaced nipples, and lymphedema of the hands and feet. Congenital renal and cardiovascular defects and diabetes may occur (
6). Typically, females with Turner syndrome have normal intelligence but may display specific neuropsychological deficits, including visuospatial processing problems, anxiety, impairment in social cognition, and motor functioning problems (
7). Moreover, several studies have highlighted an increased rate of attention deficit hyperactivity disorder (ADHD), ASD, schizophrenia, and depression in various Turner syndrome cohorts compared with the nonaffected female population (
8,
9). A robust literature supports evidence of an increased susceptibility to psychiatric disorders in X chromosome aneuploidies, although the pathogenic mechanisms remain largely unknown (
8,
9).
Here, we report a case of a 15-year-old girl with both mosaic Turner syndrome and a de novo mutation in the SHANK3 gene who presented with mild intellectual disability and psychiatric features.
Case Report
A 15-year-old girl presented with acute delirium in the context of mild to moderate intellectual disability and congenital nystagmus.
The patient, an only child and proband, was born to healthy and nonconsanguineous parents of French-Canadian ancestry. Maternal family history was remarkable for two of the patient’s first cousins with ADHD and an aunt with anxiety disorder, and the patient’s mother had a first cousin with mild intellectual disability. On the paternal side, the patient had a first cousin with depression and anxiety, and the patient’s father had a first cousin who committed suicide at age 40.
The patient was born at 39 weeks and 4 days by vaginal delivery after an uneventful pregnancy. Her birth weight was 3,465 g (73th centile), and her length was 51 cm (82th centile). Her Apgar score was 5–8, since she experienced respiratory distress at birth as a result of meconium aspiration, which promptly resolved. She was diagnosed with pendular nystagmus during the first year of life.
In terms of development, the patient was able to walk at 18 months and said her first words around age 2. She was found to have fine motor and language delay, for which she received speech and occupational therapy. At age 8, she was diagnosed with mild to moderate intellectual disability and was found to be in the spectrum of ADHD and anxiety disorders. The patient’s childhood was remarkable for lower limb heterometria (left leg >2 cm than the right leg) and left grade I hydronephrosis that progressively resolved. Her parents reported recurrent episodes of rash (mainly on the patient’s face), with features of photosensitivity. The patient had her first menstrual cycle at age 13 and regular periods afterward.
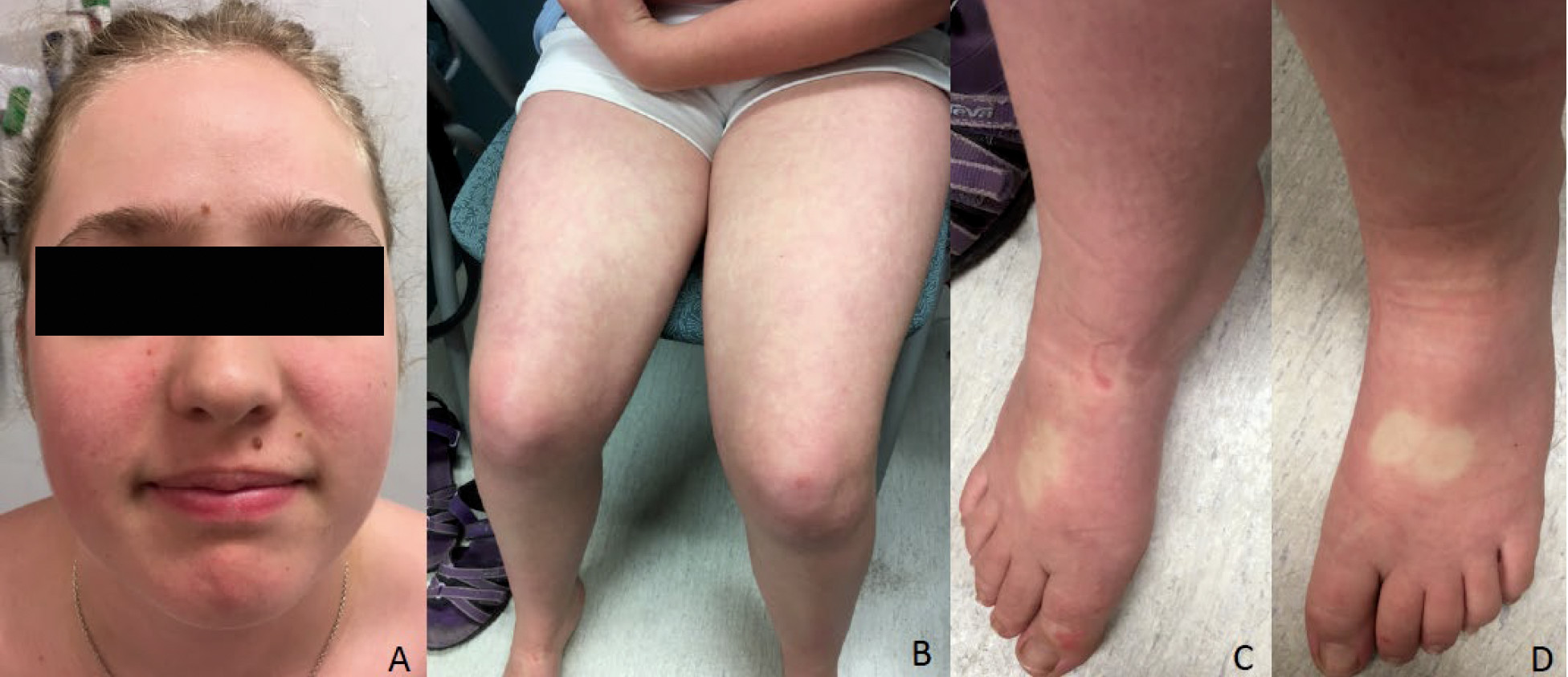
At age 15, she was admitted to the hospital in a confusional state. Neuropsychiatric evaluation revealed anxiety, sleep deprivation, racing thoughts, and visual hallucinations. A diagnosis of delirium was determined, and the patient was promptly started on a low dosage of antipsychotic medication and showed gradual treatment response in the following weeks. Neurological assessment was grossly normal, although the patient exhibited poor coordination. Her growth parameters were in the normal range (weight, 51 kg [37th percentile]; height, 154 cm [10th percentile]; occipitofrontal head circumference, 54.3 cm [50th percentile]). Minor dysmorphisms were observed, including high forehead, clinodactyly of the fifth finger bilaterally, and apparent low-set nipples. Mild pitting edema in the patient’s feet was also observed. She presented with a rash, mostly on her face and limbs, suggestive of flushing with on-off appearance (
Figure 1). Results from an extensive metabolic work-up, including urine porphobilinogen, porphyrin excretion, and sialic acid, were normal. Toxicological and immunological screening yielded negative results. Brain MRI and EEG did not show intracranial abnormalities or epileptic activity. Array comparative genomic hybridization revealed a mosaic deletion of X chromosome in 20% of the analyzed cells, consistent with Turner syndrome mosaicism. High-resolution karyotyping confirmed this diagnosis (45×[5]/46,XX[25]).
An intellectual disability and ASD panel (GeneDx, Gaithersburg, Md.), including about 2,500 genes related to intellectual disability and ASD, revealed a de novo heterozygous variant (c.2434_2449del16,p.G812RfsX76) in SHANK3 (NM 033517.1). This variant, never reported previously in the literature, causes a frameshift starting with codon glycine 812, changing this amino acid to an arginine residue, thereby creating a premature stop at position 76 of the new reading frame.
A similar episode of delirium recurred a month later, potentially in the context of appendicitis. The delirium was treated with a slightly higher dose of medication, with satisfactory response, especially with regard to anxiety and regularization of sleep pattern. However, at the 6-month follow-up the patient continued to experience some residual deficits in executive functions, particularly with regard to working memory, initiation, planning, and organization of complex tasks.
Discussion
On the basis of the existing literature, it is likely that both
SHANK3 mutations and Turner syndrome mosaicism played a role in the above patient’s psychiatric presentation, along a threshold model of multifactorial inheritance (
10).
Recently, increased literature has outlined a crucial role of
SHANK3 mutations in a broad range of neuropsychiatric disorders, including ASD, schizophrenia, mood instability, delirium, psychosis, bipolar disorder, and cognitive regression (
1,
3,
11). Several functional studies have provided insight into the pathomechanism of
SHANK3 at the postsynaptic density. Shank3 mutant mice show dysfunction of synaptic transmission, displaying self-injurious behavior, and deficits in social interaction and communication (
12,
13). Interestingly, Shank3 protein has been shown to be downregulated in Alzheimer’s disease, ASD, Phelan-McDermid syndrome, bipolar disorder, and schizophrenia, suggesting that a deficit in Shank3 expression is a common denominator of a wide range of neurodevelopmental disorders exhibiting synaptic disorganization (
12). Each of the three different Shank3 isoforms has been thought to have a specific role in neurites and synaptic endings according to different timing of expression, cell type, subcellular localization, and course of aging (
1,
14).
Shank3 protein consists of several domains: the Shank/ProSAP N-terminal domain, ankyrin repeats, the SH3 domain, the PDZ domain, a proline-rich region (PolyPro), and the sterile alpha motif domain. All regions are able to bind and interact with a wide range of proteins (i.e., modulators of small GTPases, actin-binding proteins, and actin modulators), unceasingly regulating vesicle trafficking and synaptic plasticity. Defects in this key cellular network have been shown to lead to dysregulation of spine morphogenesis and synaptic signaling, resulting in several neuropsychiatric disorders (
2).
To date, heterozygous missense, nonsense, frameshift, microdeletions, and splice-site pathogenic variants have been reported along the entire
SHANK3 gene, without a clear genotype-phenotype correlation (
1,
11). Our patient carries a novel variant (c.2434_2449del16), which creates a new reading frame at position 812 in the PolyPro region, resulting in a premature stop 76 amino acids downstream (p.G812RfsX76). This variant is predicted to cause loss of normal protein function either through nonsense-mediated mRNA decay or protein truncation, then losing at least the PolyPro region and the c-terminal sterile alpha motif domain, responsible for postsynaptic localization of the Shank3 protein (
2,
15). Accordingly, this pathogenic variant perfectly fits with the neuropsychiatric phenotype of our patient.
Nonetheless, the finding of mosaic Turner syndrome raised the question whether it might contribute to the phenotype of our patient, although she did not exhibit the typical hallmarks of Turner syndrome on physical examination. More specifically, a possible role of X monosomy was suspected with regard to the neuropsychiatric presentation of our patient. Indeed, females with Turner syndrome have been found to have an increased risk of developing ADHD (25%−50%), ASD (5%), and depression (up to 41%) (
9). Interestingly, Turner syndrome has been shown to be threefold more prevalent among females with schizophrenia compared with females in the general population, occurring mostly in the mosaic state (
16–
21). However, this association remains controversial, because large studies have not found significant statistical associations between Turner syndrome and schizophrenia (
17). A possible age bias in some studies has been suggested, because it is known that sex chromosome aneuploidy increases with age in peripheral lymphocyte cultures. A study of patients with schizophrenia and sex- and age-matched control subjects did not reveal significant differences of X chromosome loss between the schizophrenia and control groups (
17); a significant association has been found only in a cohort of individuals ages 40–49 years old (
22). This apparent inconsistency is likely related to the complex etiology of psychiatric disorders that results from multiple interacting genetic and environmental factors (
23).
To date, evidence of several psychiatric disorders among individuals with sex chromosome aneuploidies, such as 45X0, 46XXY, and 46XXX, has led to the identification of several loci of susceptibility over many sex chromosome regions (
8,
9), yet the mechanism of the X chromosome aneuploidy inducing psychiatric features remains largely unknown.
In females, only one X chromosome is active, because the other is inactivated randomly by the X chromosome inactivation center to ensure the same X chromosome dosage balance between females and males (
24). However, a great variability exists, because X chromosome inactivation can be nonrandom or skewed, defined by at least 80% preferential inactivation of X chromosomes, and may vary across different tissues and throughout the lifespan. About 15% of genes located in the pseudoautosomal regions (PAR1 and PAR2) on the X chromosome escape X-inactivation, because they are homologous to nucleotide sequences on the Y chromosome. According to the classic view of “gene dosage effect,” haploinsufficiency of various genes in both autosomal and pseudoautosomal regions has been suggested to be responsible for the neuropsychiatric features observed in X chromosome aneuploidies (
8,
9), without reaching clear conclusions. The fact that most reported cases of Turner syndrome with schizophrenia include a mosaic karyotype (45,X/46,XX) raises the possibility of a gene-dose effect, which may cause abnormal expression of an X chromosome gene product, leading in turn to schizophrenia susceptibility (
16,
19,
21).
Over the past decade, several studies have provided evidence that the X chromosome inactivation center by itself may confer susceptibility to neurodevelopmental and psychiatric disorders, giving rise to the sex chromosome inactive hypothesis (
8). It is noteworthy that our patient had congenital nystagmus, which has been reported in 4% of individuals in a Turner syndrome cohort (
25). Because two main genes responsible for congenital nystagmus, namely
FRMD7 and
GPR143, are located on chromosome X, it is possible that their haploinsufficiency, if combined with skewed X-inactivation, is responsible for the nystagmus in these patients. Interestingly, increased X chromosome inactivation skewedness has been shown in females with ASD and intellectual disability (
26), and overexpression of the
XIST gene, a master in X chromosome inactivation control, was found in lymphoblastoid cells in female patients with depression and bipolar disorder (
27). Altogether, we might speculate that an increased susceptibility to schizophrenia in mosaic Turner syndrome may, at least in part, arise from an abnormal expression of multiple gene networks, directly or indirectly controlled by the X chromosome inactivation center, resulting in an abnormal gene-dosage effect between 46XX and 45X0 cells.
Currently, there is a growing consensus that Turner syndrome phenotype is not merely caused by a genomic imbalance from deleted genes but may result from epigenetic changes in specific downstream target genes, even on autosomal chromosomes, triggered by the total or partial absence of one sex chromosome (
28). In this view, we should be mindful of the possible role of tissue-specific epigenomic and transcriptomic alterations in the brain of 45X females, similar to that observed in nervous tissue in individuals with the 46XXY karyotype (
29).
In conclusion, we report a novel SHANK3 mutation in a 46/45X0 female presenting with intellectual disability and psychiatric features. We have reviewed the current literature pertaining to psychiatric presentation in SHANK3-related disorders and Turner syndrome and suggest a possible combined effect in our patient. Whereas the pathogenic role of SHANK3 is well defined, further studies are needed to shed light on a link between Turner syndrome and psychiatric disorders. Our findings suggest that in patients with psychiatric disorders and Turner syndrome, the possibility of a second genetic hit should be explored during clinical genetic evaluation of the patient. Additionally, this case highlights that some patients admitted to the hospital for psychiatric symptoms would benefit from clinical genetic evaluation.