Persons with traumatic brain injury (TBI) are at increased risk for neuropsychiatric symptoms in the subacute and chronic postinjury periods (
1–
3). Emotional and behavioral dyscontrol, including posttraumatic irritability (
4–
7), agitation (
8), and associated neuropsychiatric symptoms occur commonly (
2). In contrast to manic and hypomanic episodes, in which irritability is excessive and persistent (
9), posttraumatic irritability reflects disturbances of moment-to-moment emotional experience and expression (
10). That is, persons with posttraumatic irritability experience transient, excessively intense, and contextually inappropriate anger outbursts but are not persistently and excessively irritable between these brief paroxysms of emotion.
In the acute to subacute period after TBI, posttraumatic irritability may arise in response to nearly any stressor or frustration (
11). Similar to other disorders of affect, such as affective lability in the early postinjury period, posttraumatic irritability may resolve during the first year postinjury (
11). This course of recovery is characteristic of persons with uncomplicated mild TBI (
4).
Among those with complicated mild TBI, moderate TBI, or severe TBI, posttraumatic irritability may become a chronic problem, with rates as high as 35%−71% (
12,
13) across these injury severities; the highest rates are found among those with the most severe injuries. As a chronic problem, posttraumatic irritability presents with recurrent, transient, ego-dystonic outbursts that may be triggered by emotional events that range from salient to affectively trivial events. These paroxysms of anger represent a distinct change from preinjury affective responses and often are regarded by the person with the injury, family, and others as being out of character (
10,
11). Posttraumatic irritability can occur with other symptoms of posttraumatic emotional and behavioral dyscontrol, including affective lability, agitation, verbal outbursts, and aggression (
2,
4,
8,
12,
14).
Two of the authors of the present study (TPB, DBA) reported reductions in posttraumatic irritability among persons with TBI who attended an alcohol use disorder clinic (
15). Treated clinically with valproate (N=14) or carbamazepine (N=4) in this uncontrolled case series, both posttraumatic irritability and alcohol use frequencies dropped significantly. This led the investigators to ask whether problem drinking among persons with posttraumatic irritability might be a form of self-treatment. The present study therefore sought to evaluate the efficacy of divalproex sodium (VPA) on posttraumatic irritability among persons with TBI and comorbid alcohol use disorder. We hypothesized that that VPA treatment would reduce the severity of posttraumatic irritability and result in lessened alcohol drinking.
Methods
Human Research Protections and Registrations
This study was approved by the Colorado Multiple Institutional Review Board and by the U.S. Army Medical Research and Materiel Command Office of Research Protections, Human Research Protection Office. All participants provided written informed consent for study participation and provided written releases for investigator contact with a knowledgeable informant capable of providing corroborative reports on data gathered through participant interview.
Study Design
This study was a prospective, randomized, double-blind placebo-controlled clinical trial. Participants were enrolled during the period from January 2010 through August 2014 at the Eastern Colorado Health Care System VA Medical Center in Denver, Colorado.
Participants were recruited using local media as well as through clinical referral networks within the academic medical center and community in which the study was performed.
Participants
Inclusion criteria.
Individuals eligible for study participation were capable of providing independent informed consent for research participation; were able to speak, read, and understand English at a level required for consent and valid completion of study measures; were 18–64 years old at the time of study enrollment; had a history of self-reported nonpenetrating TBI, using the case definition of TBI offered by the American Congress on Rehabilitation Medicine (
16) to classify participants as having mild or moderate—that is, more than mild—TBI at least 1 year prior to study participation; were experiencing functionally limiting posttraumatic irritability as reported both by the participant and a knowledgeable informant; demonstrated concordance on participant and knowledgeable informant ratings of irritability and associated symptoms on at least three of the study-relevant items of the Agitated Behavior Scale (ABS) (
17) listed below; met DSM-IV-TR criteria for alcohol abuse or dependence; and female participants had a negative pregnancy test and agreed to use barrier contraception during study participation to reduce the risk of fetal valproate exposure.
The study-relevant ABS items by scale number were as follows: 1=short attention span, easy distractibility, and inability to concentrate; 2=impulsive, impatient, and low tolerance for pain or frustration; 3=uncooperative, resistant to care, and demanding; 4=violent or threatening violence toward people or property; 9=restlessness, pacing, and excessive movement; 11=rapid, loud, or excessive talking; 12=sudden changes of mood; and 13=easily irritated or excessive crying or laughter.
Exclusion criteria.
Individuals were excluded from study participation when study assessments identified any of the following: penetrating TBI or TBI requiring neurosurgical intervention; stroke; dementia (major neurocognitive disorder) due to any cause; alcohol amnestic syndrome; seizures other than those associated with alcohol withdrawal; current or lifetime history of psychosis, pre-TBI bipolar disorder, or anxiety disorder as identified by clinical history and the Mini-International Neuropsychiatric Interview (MINI) 5.0.0 (
18); suicidal or homicidal ideation; active liver disease; symptomatic thiamine, folate, or vitamin B-12 deficiency; medical conditions that would contraindicate treatment with VPA, including known hypersensitivity to this medication, clinically significant hepatic or renal impairment, thrombocytopenia or other significant hematologic abnormalities, or concomitant medications with known interactions with VPA (e.g., substrates for CYP2C, glucuronyl transferase, and epoxide hydrolase); abnormalities on magnetic resonance imaging of the brain consistent with causes other than TBI when located in areas involved in emotion generation or regulation or requiring neurosurgical intervention (e.g., acute or chronic intracranial hematomas).
Study Assessments
Telephone interviews established the history and relative severity of TBI, as well as substance use history, posttraumatic irritability and associated neuropsychiatric symptoms, medical and psychiatric histories, and the availability of a knowledgeable informant—that is, a person with frequent face-to-face contact with the participant, usually a spouse or family member. Participants meeting study eligibility criteria based on telephone screening were invited to participate in the in-person study assessment.
Prior to any in-person screening procedures, written informed consent for study participation was obtained. In addition to confirming eligibility information acquired during telephone screening, mental health, substance use, physical health, pain, and sleep histories were assessed using validated instruments. Screening cognitive assessments included Mini-Mental State Examination (
19) and Delirium Rating Scale (
20) and required normal performance on these measures for study eligibility.
Thereafter, assessment of potentially eligible study participants also included the MINI, version 5.0.0 (
18), Beck Scale for Suicide Ideation (BSSI) (
21), Time Line Follow-Back for Drugs and Alcohol (TLFB-DA) (
22), and Neurobehavioral Rating Scale-Revised (NRS-R) assessing cognition, emotional disturbances, positive and negative symptoms, and oral/motor symptoms (
23); Trail-Making Test, Part B (TMT-B) (
24) and Frontal Assessment Battery (FAB) (
25) assessing executive cognition; physical examination, complete blood count and liver function tests; breath ethanol analysis; Clinical Global Impression–Self (CGI-S) self-report; and Clinical Global Impression–Observer (CGI-O) of study staff impressions of participants.
Interviews of the knowledgeable informants about posttraumatic irritability occurred either in person or by telephone and focused on the study-relevant ABS items (
17) noted above.
Treatment Allocation
Following randomized treatment allocation using random number generation and assignment by the hospital’s research pharmacy, trained study staff conducted 10 weekly interviews with participants and five biweekly interviews with their knowledgeable informants. Assessment of study participants during these weekly interviews included the study-relevant items on the ABS, NRS-R, BSSI, TLFB-DA and breath ethanol concentration, adverse event assessments, CGI-S, and CGI-O.
Participants allocated to double-blind treatment with VPA received a blinded 250-mg test dose. Both active drug– and placebo-assigned participants were assessed for adverse events by the blinded principal investigator. In the absence of adverse events in response to the test dose of VPA, the randomized active drug participants received VPA doses in an individualized titration protocol over the first study week, to achieve serum valproate levels between 50 and 100 mcg/ml. After achieving those levels, treatment was continued through study weeks 2–8, with blinded dose adjustments as needed to maintain serum valproate level in the 50–100 mcg/ml range, followed by a taper to treatment discontinuation by study week 10.
Neither the principal investigator nor the study staff had access to the blood level data. Only the senior research pharmacist and the study safety physician monitored dose and blood level assessments as well as side effect reports from the blinded study staff for both participant safety and accuracy of medication administration. The study safety physician authorized dosage changes that were filled by the research pharmacist and provided to the study participants in coded containers to which the study staff remained blind.
Active drug and placebo capsules were identical and prepared by the dedicated research pharmacy. Participants allocated to placebo treatment underwent identical dosing-related procedures and weekly blood tests to maintain allocation concealment.
Statistical Approach
A mixed-effects model was used in this study, which not only is a robust analytical tool with respect to missing data but also establishes an intercept reflecting individual differences at baseline. Baseline data were analyzed to determine whether there were any differences between the treatment and control groups (
Table 1). Outcome data from active study drug versus placebo data gathered in weeks 2 through 8 were analyzed for each outcome measure. Analysis began at week 2, allowing for acclimation to VPA maximal effect at steady state. For each measure, an estimated mean score was derived for each group. To control for the repeated measurements of the data, the mixed-effects model was employed.
In order to identify potential postrandomization imbalances, treatment groups were compared using chi-square tests for categorical variables, such as gender, and t tests for continuous variables, such as baseline NRS-R scores. Factors that appeared unbalanced were considered for their potential impact on the outcome and entered as control variables in sensitivity analyses of the main findings. While adding further covariates strained the analytic power, due to the modest sample size, comparing the possible covariates across treatment revealed no obvious differences. Therefore, while it is possible that various covariates might have an impact on outcomes, the balanced arms suggest the impact on these results is minimal.
Differences in efficacy between the two groups were tested using a longitudinal mixed-effects model, controlling for repeated measurements using a random intercept. Statistical significance (p<0.05) of the treatment group main effect was interpreted as an unambiguous efficacy signal, while statistical significance of the group-by-week interaction indicated a different pattern of treatment response over time between the two treatment groups. The mixed-effects model is robust to data missing at random where any differences in the observed and missing outcomes can be explained by observed values such as treatment group assignment.
The analysis included all participants who were randomly assigned to either treatment arm and received the minimal amount of treatment, being titrated to initial valproate levels. Although participants had varying frequencies of follow-up, the mixed-model analysis incorporated available data while adjusting for any kind of missingness other than informative missingness (
26). Participants’ data were implicitly weighted by response frequency, so that those with less follow-up contributed less weight to the ultimate outcomes.
Results
Sample Selection and Characteristics
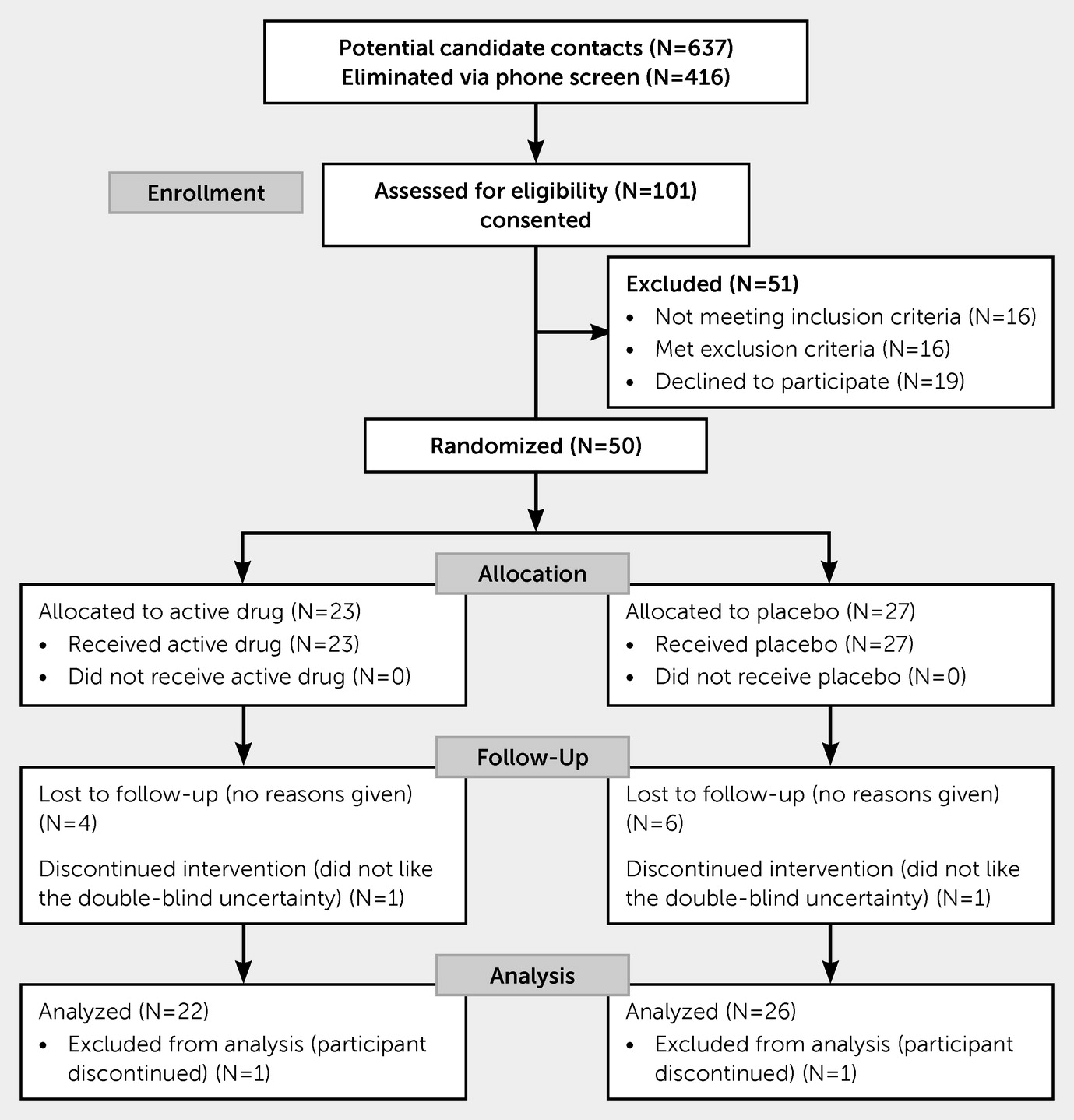
Of the 637 participants screened by telephone, 101 met initial eligibility criteria and were screened in person (
Figure 1). Of these 101 potentially eligible participants, 16 did not meet inclusion criteria, 16 met exclusion criteria, and 19 opted not to participate. A total of 50 eligible participants (40 men, 10 women) were randomized to VPA (N=23) or placebo (N=27) groups. Of those, two subjects withdrew within the first week. Eleven did not complete the full trial; these were evenly split between VPA (N=6) and placebo (N=5). None of the subjects were lost due to discontinuation of the VPA or placebo, and there were no reports of fatigue or lessened energy. A total of 37 subjects completed the trial, and data for 48 subjects were available for analyses.
There were no baseline differences between the two groups with respect to age or gender, estimated severity of TBI or of posttraumatic irritability, either on the full ABS or the 8-item ABS subscale. Likewise, there were no group differences in observed neurobehavioral disturbances (NRS-R) either on the total scores or on the mood/affect subscale, or the subjects’ own ratings of global severity of symptoms (CGI-S). There was a statistical difference in provider ratings of the CGI-O—on average, a 0.7-point difference in this 7-point scale that we judged clinically insignificant in sample characterization. There were no between-group differences on tests of executive function (TMT-B, FAB) or in the reported number of drinks per week consumed.
Study intake identified 48 proxy relationships for use in posttraumatic irritability ratings. These included spouse (26%), significant other (20%), friend (18%), parent (12%), sibling (12%), adult child/stepchild (4%), roommate (2%), or counselor (4%). Of those identified at the outset, 39 (78%) made themselves available for scheduled telephone contact in weeks 2, 4, 6, 8, and 10 for a possible total of five interviews per person through the treatment trial. On average, each identified proxy person completed 3.21 (SD=0.98) ratings.
The average daily dose of the VPA at steady state in week 2 was 963 mg (SD=122), with a range of 750 mg to 1,250 mg, dosed in 250-mg increments. This dose remained relatively stable with averages of 1,014 mg (SD=201) and 1,071 mg (SD=206) daily at weeks 3 and 5, respectively, while doses continued in the 750-mg to 1,250-mg range as titrated to the blood VPA levels.
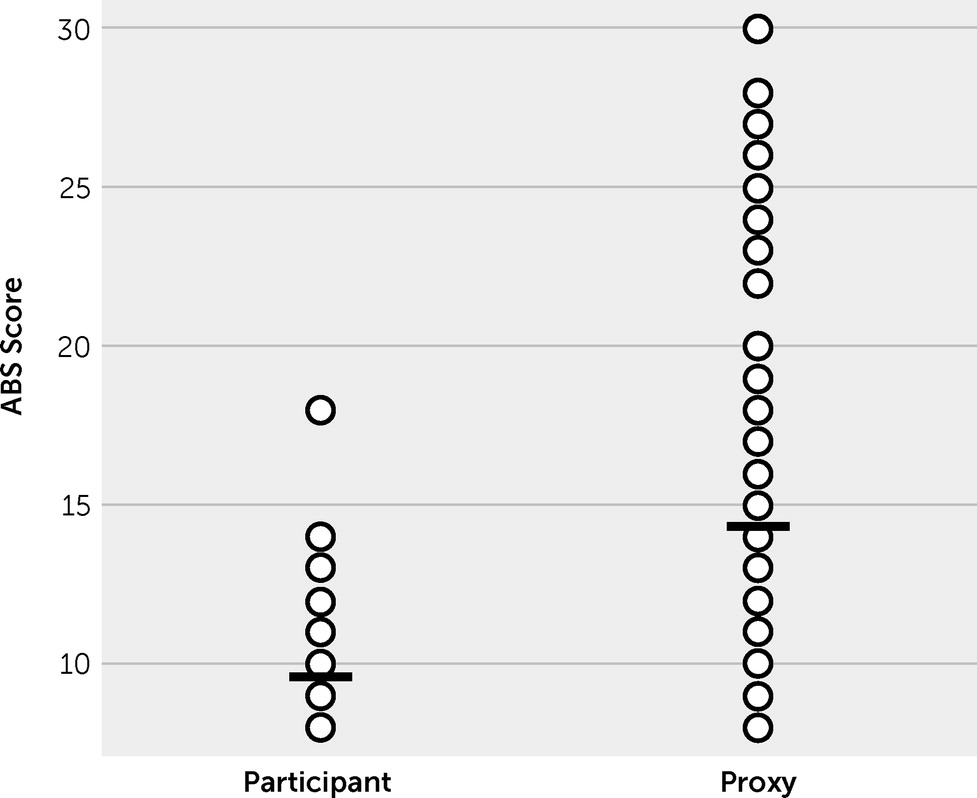
Comparing participants’ versus proxies’ paired ratings of participants’ posttraumatic irritability at treatment onset (week 2) revealed a strong discordance in perceived symptom severity (
Figure 2). The participants on average viewed their posttraumatic irritability (9.5 [SD=2.5]) as less severe than did their knowledgeable informants (14.8 [SD=3.2], p<0.05).
Posttraumatic Irritability
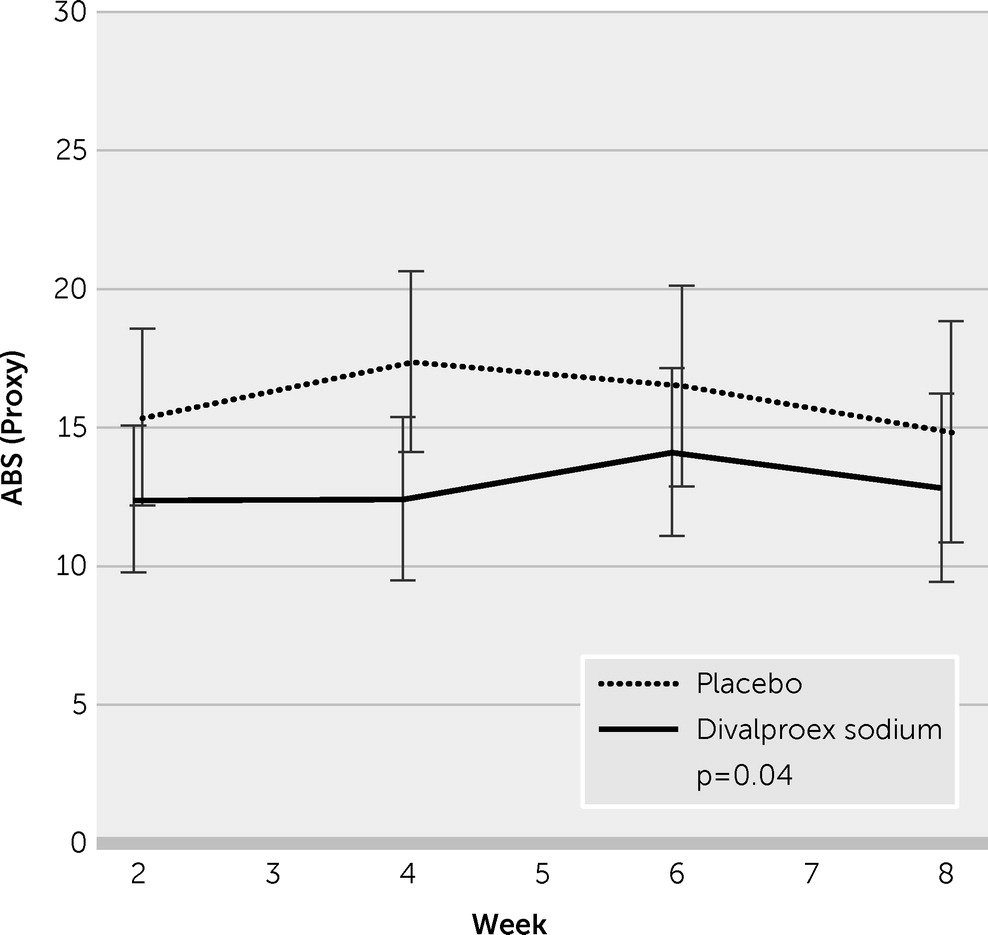
Table 2 lists mean outcomes by treatment group at week 8, adjusted for repeated measures. Mean ratings of the subjects’ posttraumatic irritability by their proxy informants on the eight ABS items demonstrated significant lowering, reflecting less severity, in the VPA group when compared with the placebo group (12.72 [SE=1.09] versus 16.25 [SE=1.26], p=0.04). As shown in
Figure 3, the VPA effect was seen in week 2 and persisted through the 8-week trial. Assessed as biweekly averages during the prospective drug trial, the mean differences reached stronger statistical significance (p=0.03; Cohen’s d=0.44).
Alcohol Use
Treatment with VPA did not change the quantity and frequency of ethanol use significantly (
Table 2).
Veteran Status
The study randomization procedure equally distributed participants by veteran/nonveteran status to the treatment and placebo groups respectively. There were no statistically significant effects of veteran status on any of the outcomes.
Anxiety
NRS-R anxiety did not differ between treatment groups (p=0.14) or change over time (p=0.53). Similarly, positive MINI diagnosis frequencies for generalized anxiety disorder (GAD; N=25, 52%) had no effect on outcome either at 2 weeks (p=0.80) or at 8 weeks (p=0.90). Chi-square test did not separate the MINI-derived posttraumatic stress disorder (PTSD) and GAD diagnoses (p=0.44)
Posttraumatic Stress Disorder
Nineteen (38%) of the subjects endorsed PTSD syndrome. There was no effect of treatment on MINI-assessed PTSD symptoms.
Safety and Tolerability
The study collected data from the three study instruments that bear on the potentially sedating effects of VPA: NRS-R, TMT-B, and FAB scores. Two NRS-R factors noted in previous investigations of persons with TBI appeared particularly relevant to the evaluation of sedative and cognitive effects: intentional behavior and memory (Factor I) and lowering of arousal state and attention (Factor IV) (
27). There were no observed effects and no adverse effects on NRS-R scores, including Factors I and IV, or on FAB or TMT-B scores.
With regard to other clinical side effects, two subjects (one each in the drug and placebo groups) reported an increase in weight that was controlled by attention to diet.
Discussion
Treatment with VPA provided a significant and sustained reduction in the severity of posttraumatic irritability, as assessed by knowledgeable informants, in this cohort of persons with chronic TBI and comorbid alcohol use disorder. The treatment group demonstrated a 20% reduction in posttraumatic irritability scores when compared with the placebo group. The between-group difference at treatment outcome yields a Cohen’s d of 0.44, a medium effect size (
28).
The study participants rated their posttraumatic irritability as less severe at baseline than did their knowledgeable informants and rated their VPA responses as not significantly different at week 8. Whereas other studies of TBI note similar discrepancies between patient and informant reports (
29,
30) and appear most problematic in the context of relatively severe TBI, factors accounting for the difference in perceived symptom severity were not studied here. The present observations suggest that accurate assessment of posttraumatic irritability and the effects of treatment may require an informant in measuring the everyday functioning of persons with posttraumatic irritability who participate in studies such as this one.
The beneficial effects of VPA did not extend to anxiety, either generalized or in PTSD. Clinician-administered assessments, including the NRS-R and the MINI, formed the basis of these observations. In future studies, the effects of VPA on co-occurring anxiety symptoms should include clinician-administered, self-report, and informant-report assessments.
While most of the subjects in this study were veterans (60%), many were not (40%). This allowed comparison of VPA effect on posttraumatic irritability in relation to veteran versus nonveteran status, a comparison that revealed no differences in treatment efficacy. This suggests that posttraumatic irritability merits treatment consideration irrespective of veteran status.
Possible Mechanisms of Action of VPA on Posttraumatic Irritability
The pharmacology of VPA remains incompletely understood despite its availability for clinical use during the past several decades. Possible mechanisms include enhanced GABAergic function. VPA increases GABAergic signaling through a series of pre- and postsynaptic mechanisms (
31,
32). For example, along with increased GABA levels, VPA causes increased GABA release and enhanced responses at the GABA receptors, prolonging the decay time of the inhibitory response of GABA
A (
33) and increasing binding of GABA
B (
34,
35). VPA may also exert its therapeutic effects on posttraumatic irritability through modifications of glutamate, serotonin, or dopamine signaling (
31,
32). This may occur in ventral limbic and paralimbic areas driving emotion generation and expression as well as in dorsal prefrontal areas that facilitate emotion regulation.
VPA regulates gene expression through both transcription factors and epigenetic regulation. Epigenetic mechanisms regulating expression of an array of genes likely play a significant role in the therapeutic effects of VPA in functional recovery from TBI (
32,
36,
37). VPA acts as a direct histone deacetylase inhibitor that leads to a transcriptionally active form of chromatin and increased transcription (
38). Therefore, VPA may modify the expression of genes involved in the chronic recurrence of posttraumatic irritability. For example, decreased histone acetylation has been reported in animal models of TBI, suggesting that VPA may restore normal acetylation and enhance genes necessary for neuronal plasticity and recovery (
39,
40). Acute VPA administration in animal models enhances motor and cognitive function after TBI (
39,
40). However, the possibility exists that improvements in neural systems subserving emotional regulation, as in frontal networks, may contribute paradoxically to reductions in emotional outbursts, a characteristic of posttraumatic irritability.
Among the other molecular targets and cellular processes that alone or in concert might contribute to the VPA mitigation of posttraumatic irritability are neurotrophic factors. These include brain-derived neurotrophic factor that VPA upregulates, a proposed contributor to its efficacy in mood disorders (
41). This upregulation may play a putative role in any neuroprotective effects of VPA (
39,
40) and might modify the neuropathological consequences of TBI (
32,
41,
42).
Other Treatment Approaches for Posttraumatic Irritability
The treatment of chronic posttraumatic irritability includes pharmacotherapy and psychotherapy (
10). The evidence for nonpharmacologic interventions includes counseling, psychotherapy (
43), and manualized anger self-management training developed specifically for persons with TBI (
44). Additionally, interventions that concurrently focus on improving functional cognition and emotional self-regulation may improve posttraumatic irritability (
45,
46).
Selective serotonin reuptake inhibitors have received attention as first-line pharmacotherapies for posttraumatic irritability (
2). However, the evidence supporting this consists principally of case reports, case series, and inferential reasoning from the treatment of irritability in other contexts. The evidence for the treatment of posttraumatic irritability includes only three randomized controlled trials, two of amantadine (
47,
48) and another of carbamazepine (
49), which provide mixed evidence of efficacy. A single-site randomized controlled trial of amantadine treated chronic posttraumatic irritability and aggression among persons with relatively severe TBI occurring from 6 months to an average of 5 years postinjury (
47). In that study, active drug significantly reduced posttraumatic irritability and was generally well tolerated. By contrast, a randomized controlled, multicenter trial recruited participants with relatively severe TBI at least 6 months prior to study enrollment (
48). That study failed to demonstrate a significant difference between amantadine and placebo, reflecting a high placebo response rate attributed to the psychosocial support provided to all study participants. The amantadine group experienced a relatively modest additional benefit of 7% over that observed in the placebo group.
A third controlled, randomized clinical trial used carbamazepine to treat chronic irritability and aggression among persons with relatively severe TBI at least 6 or more months postinjury (
49). Treatment did not improve self-reported or informant-rated posttraumatic irritability or aggression. Again, unexpectedly robust placebo effects confounded efficacy evaluation.
These medication trials demonstrate that studies of outcome and efficacy of pharmacotherapies for posttraumatic irritability bring challenges. A best course uses multiple perspectives—including participant, informant, and clinician—along with proper management of placebo effects. The absence of an apparent effect of carbamazepine stands out in relation to the present study: The benefits brought by VPA are unlikely to represent a class effect of anticonvulsants, pointing instead to single agents.
Limitations
In addition to evaluating the efficacy of VPA on posttraumatic irritability, this study aimed to evaluate concurrent efficacy on problem drinking after TBI. This remains an important problem in this population (
50,
51), but the present data do not support an effect of VPA on drinking. However, while all of the subjects in this study met criteria for alcohol abuse or dependence, they reported light use: 0.5 to 0.7 standard drinks/day, below the established moderate drinking guidelines for adults (
52). The present findings do not exclude a possible benefit of VPA among those who drink alcohol heavily after TBI; further study of this potential treatment remains important to pursue.
The study recruited a relatively small cohort of persons with TBI based on self-reported histories without, for example, day-of-injury medical records. These study results require replication in a larger sample of persons with well-characterized TBI without concurrent alcohol use disorders, ideally in a multisite trial, before generalizing the present observations. Large study designs will permit examination of other important clinical outcomes, including the relationship of posttraumatic irritability to functional status, social connectedness, and quality of life before and after treatment with VPA.
Conclusions
In this randomized, placebo-controlled double-blind clinical trial, treatment with VPA provided a significant and sustained reduction in the severity of posttraumatic irritability, as assessed by knowledgeable informants, in this cohort of persons 1 or more years after TBI who also reported comorbid alcohol abuse or dependence. Further longitudinal clinical studies of VPA treatment of posttraumatic irritability will elucidate whether the apparent ameliorating effect of VPA changes the natural history of posttraumatic irritability for the better. Such longer term questions bear examining in current and former military service members, victims of domestic abuse, persons involved in motor vehicle accidents, participants in contact sports, and the myriad other contexts of risk for TBI.