Positron Emission Tomography: Updates on Imaging of Addiction
Publication: The Journal of Neuropsychiatry and Clinical Neurosciences
Addiction is a disorder in the brain involving a shift from controlled experimentation to a more uncontrolled and compulsive pattern despite harmful consequences (5, 12, 13). Development of addiction has been linked to alterations in the reward system that biases behavior toward continuing with maladaptive behaviors. Individuals abusing psychoactive substances commonly cycle through excessive consumption (associated with intoxication and positive reinforcement), abstinence (associated with negative affect and negative reinforcement), and craving (associated with preoccupation and anticipation) resulting in relapse. Understanding the neural basis of psychoactive substance addiction has required an integrated approach spanning from well-controlled preclinical experiments to clinical studies in cognitive and affective neuroscience (14). Most of what is known about the neurobiology of substance abuse was derived from the many preclinical animal models and the invasive mechanistic studies (e.g., autoradiography, voltammetry, microdialysis, intracerebral electrophysiology) possible in these models. Although much research has been done to understand the neurobiology in humans, the brain chemistry and the underlying mechanisms of substance addiction remain largely unknown. Prior to the development of positron emission tomography (PET) neuroimaging, most human addiction research was dominated by behavioral and pharmacological studies and assays of biomarkers from bodily fluids (e.g., blood, CSF, urine) (15). By the 1990s, studies in humans began to appear to address many of the pharmacological questions posed in preclinical studies. What has greatly aided this early-stage research is the impressive and continued development of novel PET radiotracers (2).
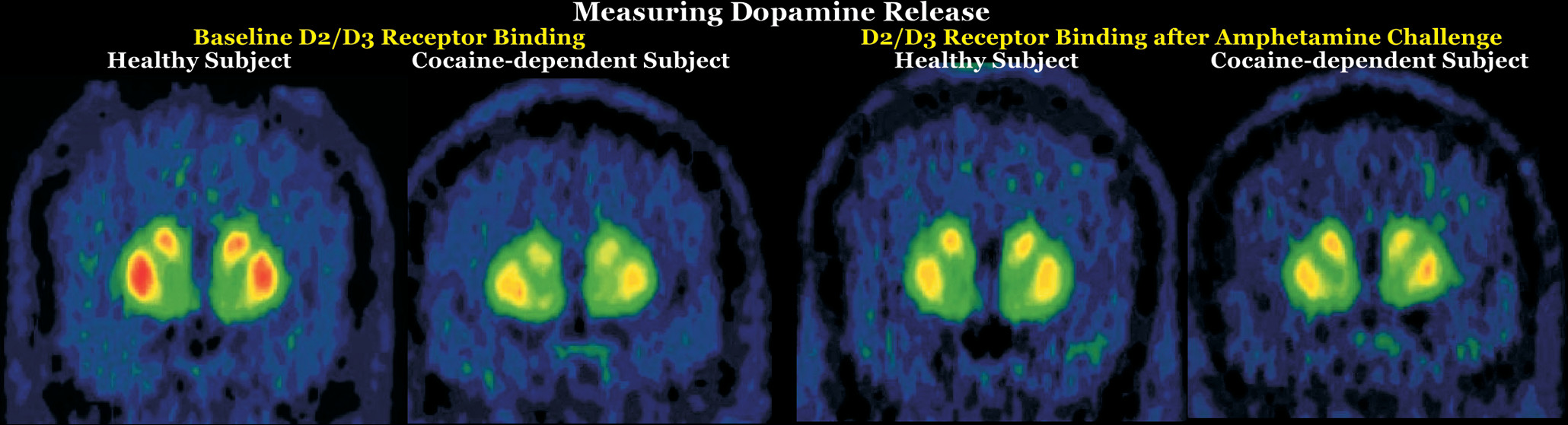
PET provides noninvasive measurements of the three-dimensional distribution of radiolabeled compounds in real-time in vivo. The ability of PET to image specific biomolecules allowing both the temporal and spatial biodistribution of a radiotracer quantitatively in vivo is unmatched. The uniqueness of PET lies in its sensitivity (nanomolar to picomolar range) and specificity. There are radiotracers available for a wide variety of molecular and cellular binding sites, such as enzymes, transporters, and receptors (Figure 1). For example, cocaine, methamphetamine, and methylphenidate were successfully radiolabeled with [11C] (half-life=20 minutes) to study the regional binding distributions of these drugs in the brain (16, 17). The short half-lives of the PET isotopes also allow repeated studies on the same subject (2, 18). Longitudinal and “test-retest” studies are powerful approaches to better understanding the role of specific targets and pharmacological interventions in treating addiction. Pharmacological displacement studies with different drugs can be measured by PET imaging studies through direct drug receptor occupancy and indirect endogenous neurotransmitter measurements during cognitive task performances. A common approach is using two consecutive PET imaging sessions in a paired-bolus paradigm, where the first (baseline) scan occurs prior to any challenge condition to assess resting radiotracer binding, while the second scan is initiated during or after the challenge (Figure 2). The challenge could compete with the radiotracer for binding sites or increase neurotransmitter signaling to compete with the radiotracer. For example, this approach was applied to measure nicotinic acetylcholine receptor (nAChR) occupancy after smoking a cigarette using a recently developed PET radioligand 2[18F]Fluoro-3-(2-S)-azetidinylmethoxy)pyridine (2FA) (19). [18F]2FA competes with administered nicotine for nAChRs. The difference between radioligand binding in the first and second scans was used to calculate nACh receptor occupancy (20). Various parameters, including background radioactivity, off-target binding, and unbound tracer binding, need to be considered to calculate the specific on-target binding potential of a radiotracer (20). Additional parameters that could affect data interpretation include distribution of receptors in brain, peripheral clearance of the radiotracer, regional cerebral blood flow, and transport of the radiotracers across the blood-brain barrier. PET has contributed greatly to our understanding of the pharmacokinetics and pharmacodynamics of drugs of abuse and has provided important information regarding drug-drug interactions, mechanisms, and toxicity, along with changes in brain chemistry.
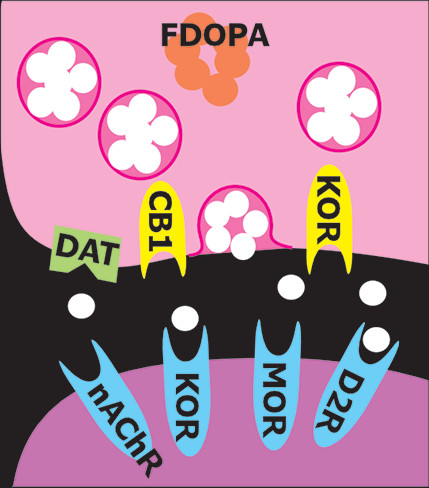
One important use of neuroimaging in addiction studies is development of biomarkers that connect what is visualized on imaging to key aspects of addiction (21). Biomarkers are vital to understanding molecular mechanisms associated with particular behavioral patterns and to improving prevention and treatment regimens of addiction. For example, one important area of research is identifying biomarkers that have potential to predict treatment response. Another main focus is identifying the neuroimaging correlates of relevant behaviors (e.g., enhanced reactivity to drug cues, poor response inhibition) and subjective experiences (e.g., cravings), since these are heavily associated with poor treatment outcomes. PET neuroimaging is an essential tool in locating the areas altered by addiction and for monitoring the effects of a treatment regimen. Studying the behavioral basis of craving may also provide insight into how addiction progresses in an individual (21).
Initial PET research on drug dependence focused primarily on dopamine-related functions due to the dopaminergic system’s central role in reward and reinforcement (1, 3, 5). Recent meta-analyses of nuclear medicine neuroimaging studies that compared healthy individuals with users to assess the effects of drugs of abuse on the striatal dopamine system have grouped drugs by whether they are stimulants (i.e., cocaine, amphetamine, methamphetamine, nicotine) or sedatives (i.e., alcohol, opioids, cannabis). However, the findings are generally similar (22–25). Lower striatal D2/D3 receptor availability was reported for individuals with amphetamine, methamphetamine, cocaine, alcohol, or opioid use (compared with healthy individuals) but not for cannabis or nicotine use (Figure 2). Lower dopamine release evoked by a challenge (i.e., blunted dopamine response) was reported for stimulant use (combining cocaine, amphetamine, and methamphetamine) and nicotine use (Figure 2). Lower dopamine transporter (DAT) availability was reported for individuals with amphetamine, methamphetamine, or opioid use (compared with healthy individuals) but not for alcohol use (mixed results for nicotine). In contrast, cocaine use was associated with the higher DAT availability. As noted in all of these meta-analyses, any of the identified differences could have been present prior to the onset of drug use (i.e., preexisting state associated with increased risk) or could have developed as adaptations to chronic exposure (22–25). Although decades of research have greatly enhanced understanding of the roles of the dopamine system in addiction, the limited therapeutic success of interventions targeting this system has increased examination of other neurotransmitter systems (3, 5).
Preclinical studies indicate that the dopamine and opioid neurotransmitter systems interact in regions associated with reward functions and that dysregulation in the mu-opioid system (which plays a key role in reward) underpins some aspects of addiction (7). Interaction between these systems in humans was recently confirmed by a PET study utilizing [11C]carfentanil to measure subcortical mu-opioid receptor (MOR) availability and 6-[18F]fluoro-L-DOPA to measure presynaptic dopamine synthesis capacity in healthy individuals and pathological gamblers (26). In both groups, presynaptic dopamine synthesis capacity was positively associated with MOR availability in the putamen, caudate nucleus, and globus pallidus. The specificity of the findings was confirmed by including a serotonin transporter ligand, [11C]MADAM, as a negative control (26). Two studies have further explored earlier findings that high MOR availability in early abstinence was associated with greater cravings and that blocking of MOR by the opiate antagonist naltrexone reduced relapse risk in a portion of patients with alcohol dependence (27, 28). One study used [11C]carfentanil, a selective MOR agonist, to investigate endogenous opioid release following dexamethasone administration in abstinent (minimum, 28 days; average, 606 days) patients with alcohol dependence and healthy individuals (28). Although baseline MOR availability did not differ between the groups, opioid release was lower in the alcohol-dependent group in half of the regions of interest (ROIs) examined, including the insula, frontal lobe, and putamen. On the basis of similar results previously reported for patients with pathological gambling, the authors suggested that opioid system dysregulation (as indicated by decreased opioid tone) may be important for both drug and behavioral addictions (28). The other study explored interactions among MOR binding, genetics, treatment response, and relapse risk in patients with alcohol dependence (27). The postmortem portion of the study found reduced MOR binding and gene expression in both striatal regions examined (ventral striatum and caudate) in individuals with heavy alcohol dependence compared with control subjects. In contrast, there were no differences in either cortical region examined (anterior cingulate and dorsolateral prefrontal cortices). The PET portion of the study was in inpatients with alcohol dependence who were about to begin a randomized clinical trial of naltrexone (27). Baseline (abstinent ≥3 weeks) MOR binding was measured with [11C]carfentanil PET. There were no significant differences between groups in abstinence rates at 90 days and at 1 year. However, a significant interaction of MOR affinity-related genotype, baseline MOR binding in striatal areas (strongest for the ventral striatum), and relapse risk was found. Particularly in OPRM1 G-allele carriers, lower striatal MOR binding was associated with a higher relapse risk. This effect was more pronounced in the naltrexone treatment group. The authors noted that for this group, higher MOR binding was associated with both higher cravings and reduced risk of relapse, suggesting that higher MOR availability might indicate patients who are more likely to benefit from naltrexone treatment (27).
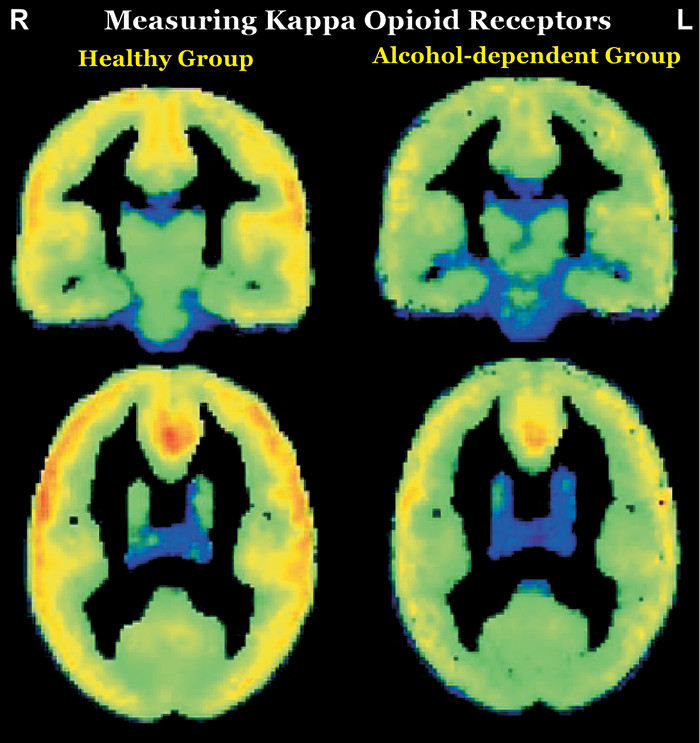
In contrast to the opioids that have abuse potential (which activate mu and delta receptors), preclinical studies utilizing opioids that activate kappa opioid receptors (KORs) have shown aversive effects (e.g., depression- and anxiety-type behaviors, dysphoria) (6–8). KOR activation decreases extracellular dopamine levels in areas that are integral to reward (i.e., ventral striatum/nucleus accumbens, dorsal striatum, medial prefrontal cortex) in part by inhibiting presynaptic release (Figure 1). Both drugs of abuse and stress upregulate the KOR system. KOR activation has been implicated in stress-induced drug seeking in preclinical studies, suggesting that blocking this receptor might have therapeutic benefit (6–8). In a recent clinical PET study, the role of the KOR system in cocaine binging and stress-induced relapse was investigated using a KOR selective agonist, [11C]GR103545 (9). Although baseline KOR binding did not differ significantly in any ROI between cocaine users (1 week of abstinence) and healthy individuals, the 3-day cocaine binge reduced KOR binding in the cocaine use group by 18% in the striatum (one of the KOR abundant areas in brain) and by 14% in the whole brain. Additionally, there was a significant positive association between baseline KOR binding and stress-induced cocaine self-administration. As noted by the authors, these findings suggest that higher KOR binding may be an indicator of greater likelihood of relapsing when stressed and that reducing KOR signaling might promote resilience in cocaine users (9). In contrast, their study utilizing the KOR selective antagonist [11C]LY2795050 to compare KOR availability between nontreatment-seeking alcohol-dependent individuals (all heavy users) and healthy individuals found that KOR availability was lower in the alcohol-dependent group by both ROI and voxel-based analyses (Figure 3) (10). They also found that reductions in drinking and craving in the alcohol-dependent group after 1 week of open-label naltrexone (100 mg daily) were negatively associated with baseline KOR binding (lower KOR availability associated with greater reductions) in several regions (ROI analysis: striatum; voxel-wise analysis: bilateral insula, bilateral cingulate cortex, and left prefrontal cortex), supporting a role for KOR in naltrexone’s therapeutic effects (11).
Preclinical studies indicate that the endocannabinoid system also interacts extensively with the dopamine system (29). Most studied is the cannabinoid1 receptor (CB1), which is a presynaptic modulatory receptor (Figure 1). CB1 agonists increase dopamine release and can induce conditioned place preference. CB1 receptors play a critical role in nicotine reward and dependence in tobacco smokers (30). A PET imaging study using the CB1 selective ligand [18F]FMPEP revealed a 20% lower total brain CB1 receptor density in tobacco smokers who were otherwise healthy compared with the nonsmoking group (30). Noting that their previous studies demonstrated similar reductions in adults with either cannabis or alcohol use disorders, the authors suggested that this may be a biomarker that is common across substance use disorders. Its role in mediating the rewarding effects of some drugs suggest that CB1 may be a promising intervention target. Cannabidiol decreases CB1 activity without inducing psychoactive effects. Both preclinical and clinical studies indicate therapeutic promise (29).
In conclusion, the diversity of PET ligands suitable for human imaging has expanded in recent years and has begun to yield a better understanding of the complex neurobiological changes underlying addiction.
There are considerable translational challenges, since clinical use will require development of safe ligands with a sufficiently long half-life for centralized manufacture and transport. When such ligands become available, if combined with the rapidly emerging field of imaging genetics and careful behavioral, cognitive, and demographic history, the field could potentially move beyond group-based analyses and into the important clinical realms of individual differences. This, in turn, has the potential to improve differential diagnosis, treatment planning, and outcome predictions. Furthermore, an important potential application for PET is identifying those at high risk for developing dependence after casual use, potentially leading to interventions early in the trajectory when it is likely to be most efficacious.
Supplementary Material
File (jnp_19080169_correction01.pdf)
- View/Download
- 78.72 KB
References
1.
van Laere K, Ceccarini J: How acute and chronic alcohol consumption modulate multiple neurotransmitter systems: a review of clinical PET neuroimaging. J Alcohol Drug Depend 2018; 6:1–6
2.
Hooker JM, Carson RE: Human positron emission tomography neuroimaging. Annu Rev Biomed Eng 2019; 21:551–581
3.
Wai JM, Levin FR, Martinez D: Neurochemical imaging in addiction: how science informs practice, in The Assessment and Treatment of Addiction, 1st ed. Edited by Danovitch I, Mooney L. Amsterdam, Elsevier, 2019, pp 1–20
4.
Trifilieff P, Martinez D: Kappa-opioid receptor signaling in the striatum as a potential modulator of dopamine transmission in cocaine dependence. Front Psychiatry 2013; 4:44
5.
Solinas M, Belujon P, Fernagut PO, et al. Dopamine and addiction: what have we learned from 40 years of research. J Neural Transm (Vienna) 2019; 126:481–516
6.
Anderson RI, Becker HC: Role of the dynorphin/kappa opioid receptor system in the motivational effects of ethanol. Alcohol Clin Exp Res 2017; 41:1402–1418
7.
Reed B, Butelman ER, Kreek MJ: Endogenous opioid system in addiction and addiction-related behaviors. Curr Opin Behav Sci 2017; 13:196–202
8.
Tejeda HA, Bonci A: Dynorphin/kappa-opioid receptor control of dopamine dynamics: Implications for negative affective states and psychiatric disorders. Brain Res 2019; 1713:91–101
9.
Martinez D, Slifstein M, Matuskey D, et al: Kappa-opioid receptors, dynorphin, and cocaine addiction: a positron emission tomography study. Neuropsychopharmacology 2019; 44:1720–1727
10.
Vijay A, Cavallo D, Goldberg A, et al: PET imaging reveals lower kappa opioid receptor availability in alcoholics but no effect of age. Neuropsychopharmacology 2018; 43:2539–2547
11.
de Laat B, Goldberg A, Shi J, et al: The kappa opioid receptor is associated with naltrexone-induced reduction of drinking and craving. Biol Psych 2019; pii: S0006-3223(19)31413-1. 2019
12.
Volkow ND, Wang G-J, Fowler JS, et al: Chapter one: neuroimaging of addiction, in Imaging of the Human Brain in Health and Disease. Edited by Seeman P, Madras B. Boston, Academic Press, 2014, pp 1–26
13.
Volkow ND, Boyle M: Neuroscience of addiction: relevance to prevention and treatment. Am J Psychiatry 2018; 175:729–740
14.
Parvaz MA, Alia-Klein N, Woicik PA, et al: Neuroimaging for drug addiction and related behaviors. Rev Neurosci 2011; 22:609–624
15.
Phelps ME, Mazziotta JC: Positron emission tomography: human brain function and biochemistry. Science 1985; 228:799–809
16.
Sai KK, Fan J, Tu Z, et al: Automated radiochemical synthesis and biodistribution of [11C]l-α-acetylmethadol ([11C]LAAM). Appl Radiat Isot 2014; 91:135–140
17.
Shumay E, Wiers CE, Shokri-Kojori E, et al: New repeat polymorphism in the AKT1 gene predicts striatal dopamine D2/D3 receptor availability and stimulant-induced dopamine release in the healthy human brain. J Neurosci 2017; 37:4982–4991
18.
Shimizu S, Hirose D, Hatanaka H, et al: Role of neuroimaging as a biomarker for neurodegenerative diseases. Front Neurol 2018; 9:265
19.
Doll F, Dolci L, Valette H, et al: Synthesis and nicotinic acetylcholine receptor in vivo binding properties of 2-fluoro-3-[2(S)-2-azetidinylmethoxy]pyridine: a new positron emission tomography ligand for nicotinic receptors. J Med Chem 1999; 42:2251–2259
20.
Kimes AS, Horti AG, London ED, et al: 2-[18F]F-A-85380: PET imaging of brain nicotinic acetylcholine receptors and whole body distribution in humans. FASEB J 2003; 17:1331–1333
21.
Garrison KA, Potenza MN: Neuroimaging and biomarkers in addiction treatment. Curr Psychiatry Rep 2014; 16:513
22.
Ashok AH, Mizuno Y, Volkow ND, et al: Association of stimulant use with dopaminergic alterations in users of cocaine, amphetamine, or methamphetamine: a systematic review and meta-analysis. JAMA Psychiatry 2017; 74:511–519
23.
Ashok AH, Mizuno Y, Howes OD: Tobacco smoking and dopaminergic function in humans: a meta-analysis of molecular imaging studies. Psychopharmacology (Berl) 2019; 236:1119–1129
24.
Kamp F, Proebstl L, Penzel N, et al: Effects of sedative drug use on the dopamine system: a systematic review and meta-analysis of in vivo neuroimaging studies. Neuropsychopharmacology 2019; 44:660–667
25.
Proebstl L, Kamp F, Manz K, et al: Effects of stimulant drug use on the dopaminergic system: a systematic review and meta-analysis of in vivo neuroimaging studies. Eur Psychiatry 2019; 59:15–24
26.
Majuri J, Joutsa J, Arponen E, et al: Dopamine synthesis capacity correlates with µ-opioid receptor availability in the human basal ganglia: a triple-tracer PET study. Neuroimage 2018; 183:1–6
27.
Hermann D, Hirth N, Reimold M, et al: Low μ-opioid receptor status in alcohol dependence identified by combined positron emission tomography and post-mortem brain analysis. Neuropsychopharmacology 2017; 42:606–614
28.
Turton S, Myers JF, Mick I, et al: Blunted endogenous opioid release following an oral dexamphetamine challenge in abstinent alcohol-dependent individuals. Mol Psychiatry 2018 (Epub ahead of print, June 25, 2018)
29.
Chye Y, Christensen E, Solowij N, et al: The endocannabinoid system and cannabidiol’s promise for the treatment of substance use disorder. Front Psychiatry 2019; 10:
30.
Hirvonen J, Zanotti-Fregonara P, Gorelick DA, et al: Decreased cannabinoid CB1 receptors in male tobacco smokers examined with positron emission tomography. Biol Psychiatry 2018; 84:715–721
Information & Authors
Information
Published In


History
Received: 1 August 2019
Accepted: 3 September 2019
Published in print: Fall 2019
Published online: 15 October 2019
Keywords
Authors
Metrics & Citations
Metrics
Citations
Export Citations
If you have the appropriate software installed, you can download article citation data to the citation manager of your choice. Simply select your manager software from the list below and click Download.
For more information or tips please see 'Downloading to a citation manager' in the Help menu.
View Options
View options
PDF/EPUB
View PDF/EPUBFigures
Tables
Media
References
References
1.
van Laere K, Ceccarini J: How acute and chronic alcohol consumption modulate multiple neurotransmitter systems: a review of clinical PET neuroimaging. J Alcohol Drug Depend 2018; 6:1–6
2.
Hooker JM, Carson RE: Human positron emission tomography neuroimaging. Annu Rev Biomed Eng 2019; 21:551–581
3.
Wai JM, Levin FR, Martinez D: Neurochemical imaging in addiction: how science informs practice, in The Assessment and Treatment of Addiction, 1st ed. Edited by Danovitch I, Mooney L. Amsterdam, Elsevier, 2019, pp 1–20
4.
Trifilieff P, Martinez D: Kappa-opioid receptor signaling in the striatum as a potential modulator of dopamine transmission in cocaine dependence. Front Psychiatry 2013; 4:44
5.
Solinas M, Belujon P, Fernagut PO, et al. Dopamine and addiction: what have we learned from 40 years of research. J Neural Transm (Vienna) 2019; 126:481–516
6.
Anderson RI, Becker HC: Role of the dynorphin/kappa opioid receptor system in the motivational effects of ethanol. Alcohol Clin Exp Res 2017; 41:1402–1418
7.
Reed B, Butelman ER, Kreek MJ: Endogenous opioid system in addiction and addiction-related behaviors. Curr Opin Behav Sci 2017; 13:196–202
8.
Tejeda HA, Bonci A: Dynorphin/kappa-opioid receptor control of dopamine dynamics: Implications for negative affective states and psychiatric disorders. Brain Res 2019; 1713:91–101
9.
Martinez D, Slifstein M, Matuskey D, et al: Kappa-opioid receptors, dynorphin, and cocaine addiction: a positron emission tomography study. Neuropsychopharmacology 2019; 44:1720–1727
10.
Vijay A, Cavallo D, Goldberg A, et al: PET imaging reveals lower kappa opioid receptor availability in alcoholics but no effect of age. Neuropsychopharmacology 2018; 43:2539–2547
11.
de Laat B, Goldberg A, Shi J, et al: The kappa opioid receptor is associated with naltrexone-induced reduction of drinking and craving. Biol Psych 2019; pii: S0006-3223(19)31413-1. 2019
12.
Volkow ND, Wang G-J, Fowler JS, et al: Chapter one: neuroimaging of addiction, in Imaging of the Human Brain in Health and Disease. Edited by Seeman P, Madras B. Boston, Academic Press, 2014, pp 1–26
13.
Volkow ND, Boyle M: Neuroscience of addiction: relevance to prevention and treatment. Am J Psychiatry 2018; 175:729–740
14.
Parvaz MA, Alia-Klein N, Woicik PA, et al: Neuroimaging for drug addiction and related behaviors. Rev Neurosci 2011; 22:609–624
15.
Phelps ME, Mazziotta JC: Positron emission tomography: human brain function and biochemistry. Science 1985; 228:799–809
16.
Sai KK, Fan J, Tu Z, et al: Automated radiochemical synthesis and biodistribution of [11C]l-α-acetylmethadol ([11C]LAAM). Appl Radiat Isot 2014; 91:135–140
17.
Shumay E, Wiers CE, Shokri-Kojori E, et al: New repeat polymorphism in the AKT1 gene predicts striatal dopamine D2/D3 receptor availability and stimulant-induced dopamine release in the healthy human brain. J Neurosci 2017; 37:4982–4991
18.
Shimizu S, Hirose D, Hatanaka H, et al: Role of neuroimaging as a biomarker for neurodegenerative diseases. Front Neurol 2018; 9:265
19.
Doll F, Dolci L, Valette H, et al: Synthesis and nicotinic acetylcholine receptor in vivo binding properties of 2-fluoro-3-[2(S)-2-azetidinylmethoxy]pyridine: a new positron emission tomography ligand for nicotinic receptors. J Med Chem 1999; 42:2251–2259
20.
Kimes AS, Horti AG, London ED, et al: 2-[18F]F-A-85380: PET imaging of brain nicotinic acetylcholine receptors and whole body distribution in humans. FASEB J 2003; 17:1331–1333
21.
Garrison KA, Potenza MN: Neuroimaging and biomarkers in addiction treatment. Curr Psychiatry Rep 2014; 16:513
22.
Ashok AH, Mizuno Y, Volkow ND, et al: Association of stimulant use with dopaminergic alterations in users of cocaine, amphetamine, or methamphetamine: a systematic review and meta-analysis. JAMA Psychiatry 2017; 74:511–519
23.
Ashok AH, Mizuno Y, Howes OD: Tobacco smoking and dopaminergic function in humans: a meta-analysis of molecular imaging studies. Psychopharmacology (Berl) 2019; 236:1119–1129
24.
Kamp F, Proebstl L, Penzel N, et al: Effects of sedative drug use on the dopamine system: a systematic review and meta-analysis of in vivo neuroimaging studies. Neuropsychopharmacology 2019; 44:660–667
25.
Proebstl L, Kamp F, Manz K, et al: Effects of stimulant drug use on the dopaminergic system: a systematic review and meta-analysis of in vivo neuroimaging studies. Eur Psychiatry 2019; 59:15–24
26.
Majuri J, Joutsa J, Arponen E, et al: Dopamine synthesis capacity correlates with µ-opioid receptor availability in the human basal ganglia: a triple-tracer PET study. Neuroimage 2018; 183:1–6
27.
Hermann D, Hirth N, Reimold M, et al: Low μ-opioid receptor status in alcohol dependence identified by combined positron emission tomography and post-mortem brain analysis. Neuropsychopharmacology 2017; 42:606–614
28.
Turton S, Myers JF, Mick I, et al: Blunted endogenous opioid release following an oral dexamphetamine challenge in abstinent alcohol-dependent individuals. Mol Psychiatry 2018 (Epub ahead of print, June 25, 2018)
29.
Chye Y, Christensen E, Solowij N, et al: The endocannabinoid system and cannabidiol’s promise for the treatment of substance use disorder. Front Psychiatry 2019; 10:
30.
Hirvonen J, Zanotti-Fregonara P, Gorelick DA, et al: Decreased cannabinoid CB1 receptors in male tobacco smokers examined with positron emission tomography. Biol Psychiatry 2018; 84:715–721