Alzheimer’s disease (AD) is characterized by progressive cognitive and functional decline and the frequent emergence of neuropsychiatric syndromes over the course of the 10- to 15-year symptomatic phase of the disease (
1,
2). AD is the most prevalent late-life neurodegenerative disorder and is becoming increasingly common as the world’s population ages. AD doubles in frequency every 5 years after the age of 60, rising from a rate of 1% among 60-year-olds to approximately 40% among 80-year-olds (
3). There are currently 5.3 million persons with AD dementia in the United States, and this is projected to rise to 14 million by 2050. The corresponding economic impact will increase from the current $230 billion annually to more than $1 trillion annually by 2050 (
4). Despite the great threat to the population posed by AD, there are limited therapies available and a high rate of failure in developing new drugs for this disorder (
5).
Two classes of cognitive-enhancing agents are currently approved for the treatment of AD and available on the market: cholinesterase inhibitors (ChE-Is; donepezil, rivastigmine, galantamine) and an N-methyl-d-aspartate (NMDA) receptor antagonist (memantine). There are currently no approved disease-modifying agents for the treatment of AD. Therapies that prevent or delay the onset, slow the progression, or improve the symptoms of AD are needed to respond to the cognitive, functional, and behavioral changes in the burgeoning AD population.
There are 121 agents currently in clinical trials for AD (
6). In this article, drug development strategies for AD and the pipeline of emerging agents for treatment of neuropsychiatric symptoms, cognitive enhancement, and disease modification are reviewed. Promising advances in the treatment of neuropsychiatric aspects of AD are emphasized.
Pharmacologic Management of Neuropsychiatric Disorders
With few exceptions—mainly involving very rare disorders—the randomized double-blind placebo-controlled trial is the gold standard for the assessment and eventual approval of therapies for all medical conditions. Clinical trials provide rigorous answers to very specific questions. They address outcomes using prespecified instruments for participants diagnosed with specific criteria, in a given stage of disease, treated for a specified period of time, with one or more doses of a test agent. Conclusions beyond these tightly specified parameters cannot be drawn from clinical trials, and trial results cannot be confidently generalized to nontrial populations.
In the practice of neuropsychiatry, treatment decisions are often not informed by the narrow outcomes of a clinical trial. Patients with mixed conditions (e.g., AD and stroke) exhibiting complex combinations of neuropsychiatric symptoms (e.g., depression, psychosis, and sleep disorder), who are older or younger than those included in a trial or have medical conditions or take medications that were not allowed in a trial, are common in neuropsychiatric practices. For this reason, off-label prescribing is frequent to achieve an improved quality of life for patients in the relentlessly patient-centric approach that characterizes neuropsychiatric practice. Neuropsychiatrists and behavioral-cognitive neurologists bear a responsibility for rational pharmacology using evidence-based medicine in a broad context. In addition to data derived from clinical trials, informative principles to guide pharmacologic decision making include the therapeutic metaphor—seeking similarities between symptoms successfully treated in clinical trials and those evident in a patient whose disorder differs from those included in the trial (e.g., use of pimavanserin for the treatment of psychosis in AD after demonstration of its utility in the psychosis of Parkinson’s disease [PD]) (
7,
8)—and the biological extension principle: that responses seen in trial participants may be recapitulated in patients with similar symptoms and a shared biology (e.g., rivastigmine was first shown to improve cognition in AD and then shown to improve cognition in PD dementia, which has a similar cholinergic deficit) (
9,
10). The greater the similarity between the trial patients and the patient to be treated, the greater the likelihood that the therapeutic extension will be successful. Case reports, multiple case observations, and well-conducted n=1 trials can provide information useful for individual patient management and may guide programs for future expanded indications of approved agents (
11,
12).
On the basis of these principles, best practices and medicine-based evidence evolve that define a body of information on which neuropsychiatric prescribers can call for guidance until clinical trials provide more specific information. Professional prescribing standards are composed of extrapolations from clinical trials, carefully observed off-label treatment responses, knowledge of potential benefit and harm, appropriate informed consent, and careful documentation in the medical record. Use of psychotropic agents in AD follow these principles since few agents have been approved by the Food and Drug Administration (FDA) for behavioral indications in AD or other neurodegenerative disorders.
Drug Development and Clinical Trials
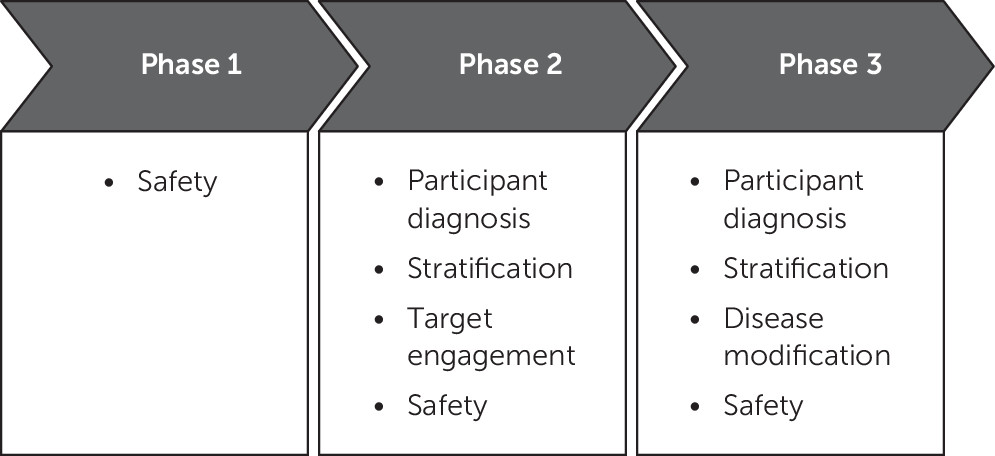
Emerging therapies typically progress from nonclinical testing of efficacy and safety in animals to human clinical trials (
24). Phase 1 consists of testing the candidate treatment in normal healthy volunteers organized into single ascending dose cohorts followed by multiple ascending dose cohorts. Safety, tolerability, pharmacokinetic parameters (e.g., bioavailability, half-life, maximum concentration, time to maximum concentration, presence of metabolites, effects of food on pharmacokinetics, drug-drug interactions with commonly used drugs, maximum tolerated dose), blood-brain barrier penetration, and doses to be advanced to phase 2 are determined in phase 1. Programs developing drugs for AD may include at least one cohort of elderly individuals to assess the effects of aging on safety, tolerability, and pharmacokinetics. A few programs include cohorts of participants with AD in mixed phase-1/2 designs. Immunotherapy studies may include only participants with AD, since immune responses could be permanently altered in healthy volunteers.
Phase 2 in AD drug development establishes proof-of-concept in participants with AD (
25,
26). Critical outcomes of phase-2 studies are determination of the doses to be advanced to phase 3; assessment of a dose-related response on clinical measurers, biomarkers, or both; demonstration of target engagement (discussed below) to establish that there is a pharmacodynamic effect in the dose range tested; and collection of additional evidence of safety and tolerability (
24). Programs for disease modifying therapies (DMTs), treatments for behavioral symptoms, and cognitive enhancement use inclusion criteria and outcomes to match their development objectives. DMT programs may accept drug-placebo differences on a biomarker as sufficient evidence of target engagement to advance a therapy to phase 3, whereas cognitive-enhancing agents and psychotropic drug development programs demonstrate efficacy on relevant cognitive or behavioral measures in phase 2 to provide a foundation for phase 3. Phase-2 trials typically involve 100–400 patients per arm, although some may be larger or smaller depending on the objectives and design of the trial.
Phase-3 trials provide confirmatory evidence of efficacy and safety (
27) and accrue data for application to the FDA for marketing approval. Trials of cognitive enhancers and DMTs of participants with AD dementia must demonstrate a drug-placebo difference on two prespecified outcomes: either cognition and a global measure or cognition and a functional measure. Cognitive outcomes address the core deficit of AD; global and functional outcomes establish the clinical meaningfulness of the intervention (
28). Candidate DMTs must show an effect on biomarkers to be regarded as disease-modifying; the magnitude of the effect and repertoire of biomarkers required are not certain, since no DMT is approved for AD. DMTs being assessed in prodromal AD can be approved by demonstrating a drug-placebo difference on a single composite measure such as the Clinical Dementia Rating–Sum of Boxes (
29) plus evidence of an effect on biomarkers (
30).
AD has a long preclinical phase that precedes the onset of AD dementia by 15–20 years. During this time, the individuals are cognitively normal but have positive amyloid positron emission tomography (PET) or an AD-type signature of biomarker changes in the CSF with decreased levels of amyloid beta-protein (Aβ) and increased levels of phosphorylated tau (p-tau) and total tau (
31,
32). In addition to individuals with abnormal state markers of AD pathology (PET or CSF), those with mutations that produce autosomal dominant AD or persons who are homozygotes for the apolipoprotein e4 gene and close to the time of onset of symptomatic AD have preclinical AD and are candidates for prevention trials (
33,
34). Secondary prevention trials are conducted during the preclinical phase of AD with the goal of preventing or delaying the onset of cognitive decline (
35).
An AD drug development program takes at least 7.6 years to execute after the agent has undergone nonclinical characterization in animals. Phase-1 trials require on average 12.8 months to complete, phase 2 takes 27.7 months on average, and phase 3 typically lasts 50.9 months (
36).
AD drug development is extraordinarily expensive. The investment cost for a single agent to reach the end of phase 1 is approximately $71 million. To the end of phase 2, the cumulative cost is $126 million, and to the end of phase 3, the investment cost is $413 million. Considering the cost of capitalization and the cost of failures, the total cost of developing a successful agent is calculated at $5.69 billion (
36). These costs are prohibitive and require that less cost-intensive strategies be developed if a robust repertoire of therapies for all aspects of AD are to be made available to patients and clinicians.
Biomarkers in Clinical Trials
The amyloid (A), tau (T), neurodegeneration (N) framework (A/T/N) identifies the core pathology of AD and specifies biomarkers for each of the components (
32) (
Table 1). For A, fibrillar/plaque amyloid is identified by amyloid PET; monomeric Aβ40 and Aβ42 can be identified in CSF; and the monomeric Aβ42/40 ratio is abnormal in the blood (
37,
38). There is no consensus on a measure of Aβ oligomers. T biomarkers include tau PET, which identifies neurofibrillary tangles; hyperphosphorylated tau (p-tau) in the CSF; and p-tau in the blood (
39). Biomarkers of N include MRI evidence of cerebral and hippocampal atrophy; fluorodeoxyglucose (FDG) PET indicative of cerebral hypometabolism; CSF total tau, neurogranin, and neurofilament light (NfL); and blood NfL (
40,
41). Biomarkers are indirect measures of cerebral pathology and have their own diffusion, metabolism, and excretion characteristics that affect their detection and usefulness as markers for therapy. They provide inferential but imperfect insight into brain pathology. Many other biomarkers are in development and promise to furnish a more comprehensive window on the pathology of AD and response to treatment (
42).
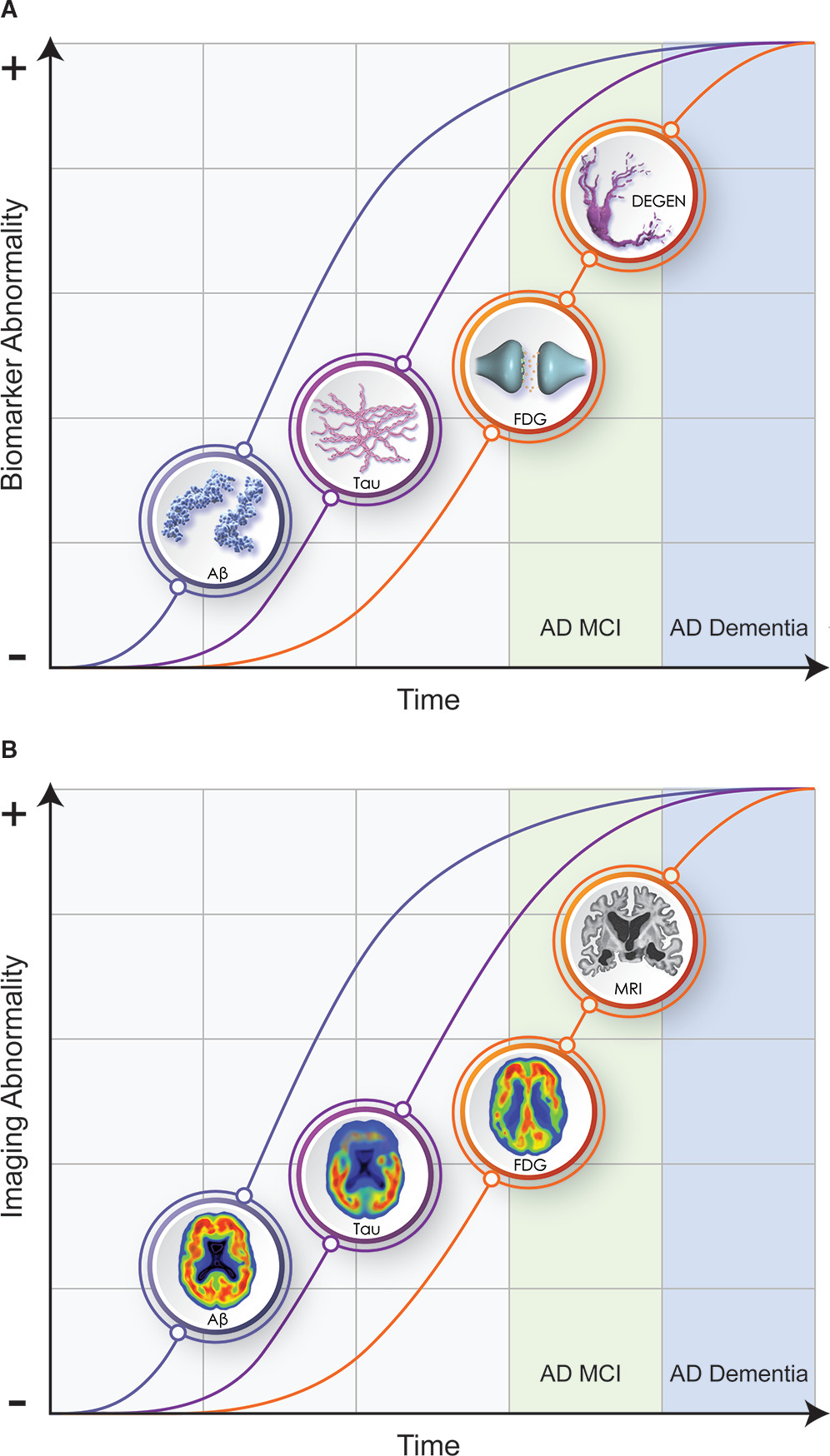
Biomarker profiles change over the course of the illness and define an AD continuum (
43,
44) (
Figure 1). Biomarker trajectories reflect the dynamic nature of the evolving pathology of AD; changes in Aβ are identifiable first (positive amyloid PET; decreased Aβ42 in the CSF), followed by alterations in CSF tau and p-tau, and in turn by cerebral atrophy on MRI and hypometabolism on FDG PET. CSF total tau and p-tau are increased early in AD and function as state markers; tau PET becomes positive later in the disease course and functions as a stage marker (
45). Cognitive and functional symptoms are relatively late manifestations, occurring coincident with evidence of T abnormalities and of N on MRI and FDG PET. Given the long course of these biomarker changes (e.g., approximately 20 years from changes in Aβ to onset of AD dementia), not all individuals with positive amyloid imaging will live to develop AD, and counseling patients on the basis of positive biomarkers must be done with caution (
46). The ability to detect biomarker changes prior to clinical abnormalities has made it possible to design secondary prevention trials in individuals at high risk for AD.
Biomarkers have transformed AD drug development (
Figure 2) (
47,
48). In phase 1, biomarkers serve as safety measures to assess adverse effects in first human trials. Hepatic injury revealed by elevated liver functions or adverse cardiac effects revealed by electrocardiography are examples of well-established phase-1 biomarkers.
In phase 2, biomarkers more specific to AD have a major role. Participant diagnosis is established by amyloid PET or CSF changes indicative of AD. Between 15% and 40% of participants referred for AD clinical trials have negative amyloid scans and are phenocopies of the disease (having the clinical phenotype of AD but not the underlying biology of AD) (
49,
50). Requiring confirmation of AD-type pathology for trial participation ensures that the target biology for the intervention is present. Participants with confirmed diagnoses decline more in the course of a trial observation period and facilitate detection of a drug-placebo difference for effective therapies (
51). Plasma Aβ 42/40 ratios may soon be able to replace amyloid imaging or CSF changes for trial entry; they could be used to determine which potential participants are likely to be amyloid positive if scanned (
37,
52,
53). The apolipoprotein E-4 (ApoE-4) genotype is a biomarker used to stratify trial populations in either the recruitment or the analytic phase of study conduct. Some biological dimensions of AD differ in ApoE-4 carriers compared with noncarriers, and genotype could influence treatment outcomes (
54). Tau PET is used to further characterize trial populations; AD participants in the preclinical period progress more rapidly to symptomatic states if they have greater tau burdens at trial baseline (
38).
Target engagement biomarkers demonstrate that pharmacodynamic mediators of the therapeutic response observed in animals are present in participants with AD. Examples of biomarkers of target engagement include showing reduced Aβ production using stable isotope labeled kinetics (
55), reduced CSF Aβ with inhibitors of Aβ-producing enzymes (
56), decreased glutaminyl cyclase enzyme activity with enzyme inhibitors (
57), and increased Aβ fragments in plasma and CSF with gamma-secretase inhibitors and modulators (
58). Drug development is currently hindered by the relative lack of availability of target engagement biomarkers.
A critical role of biomarkers in phase-3 trials is to provide evidence that supports disease modification. Of the core biological changes of AD—amyloid plaques and related amyloid species, tau pathology and neurofibrillary tangles, and neurodegeneration (
32)—neuronal loss is the final common denominator of disease progression. Demonstrating neuroprotection and drug-placebo difference in neurodegeneration is an essential aspect of supporting disease modification (
59). MRI is one approach to demonstrating a disease-modifying effect. MRI measures cerebral cortical atrophy, ventricular enlargement, and hippocampal atrophy. Loss of hippocampal volume on MRI correlates with change in hippocampal size and hippocampal neuronal number at autopsy (
60,
61). NfL and total tau in CSF and NfL in blood are biomarkers indicative of neurodegeneration (53). They have been included in relatively few trials thus far.
Assessment of safety employs biomarkers throughout drug development. Some monoclonal antibody treatments induce amyloid-related imaging abnormalities; these are detected by MRI collected serially during the trials (
62). Biomarkers convert clinical trials into precision drug development enterprises that identify potentially responsive participants, demonstrate target engagement, provide evidence of disease modification, and establish the safety of interventions (
38).
Alzheimer’s Drug Development Pipeline
We maintain a database of information derived from the federal registry ClinicalTrials.gov and review the data in the pipeline annually (
6,
63–
66). Registration on ClinicalTrials.gov is federally mandated; all sponsors—academic, governmental, biopharmaceutical industry, philanthropy—who test therapeutic agents and devices must register their studies on this site. Compliance with the mandate is high, and the database is a comprehensive summary of all ongoing clinical trials in the United States (
67–
69). Trials conducted outside the United States are not required to register on ClinicalTrials.gov. Most development programs include trials in the United States and are registered; the data available are comprehensive. The information presented here was derived from ClinicalTrials.gov as of February 27, 2020.
Trials for Treatments of Neuropsychiatric Symptoms in Alzheimer’s Disease
Substantial progress is being made in clinical trials for neuropsychiatric syndromes in AD. There are currently eight agents in clinical trials for agitation in AD: brexpiprazole, s-citalopram, lithium, dronabinol, dextromethorphan/quinidine, dextromethorphan/bupropion, mirtazapine, and prazosin (
6,
21,
70). Methylphenidate is being assessed in a trial for treatment of apathy in AD (
71). Two sleep agents—zolpidem and zolplicone—are being assessed for insomnia in AD, and one drug, lemborexant, is being studied for its effect on irregular sleep-wake rhythm disorder (
72). Pimavanserin had a successful trial for dementia-related psychosis that included AD with psychosis, PD with psychosis, dementia with Lewy bodies with psychosis, frontotemporal lobar degeneration spectrum disorders with psychosis, and vascular dementia with psychosis. Pimavanserin will be reviewed by the FDA for the indication of dementia-related psychosis (
73).
Together these AD drug development programs host 12 agents in clinical trials for neuropsychiatric disorders: none in phase 1, four in phase 2, and eight in phase 3 (
Figure 3). These observations of the drug development pipeline suggest that the repertoire of agents available for treatment of neuropsychiatric syndromes in AD and related neurodegenerative disorders will expand.
Progress in clinical trials for psychotropic agents is at least partially attributable to development of improved definitions of neuropsychiatric syndromes, including agitation, apathy, and psychosis (
74–
76). Definitions facilitate construction of more homogenous trial populations, identification of appropriate outcomes, discussions with regulatory authorities, and education of clinicians concerning appropriate prescribing. Consensus definitions facilitate nonpharmacologic research and natural history studies.
Clinical Trials for Cognitive-Enhancing Agents
Several classes of cognitive-enhancing agents have been assessed in recent clinical trials and found not to show a drug-placebo difference. These include nicotinic agents, histamine receptor antagonists,
11-β-hydroxysteroid dehydrogenase inhibitors, phosphodiesterase inhibitors, norepinephrine reuptake inhibitors, and 5-HT
6 antagonists (
13,
65,
77–
80).
Oligomannate (GV-971) was recently approved in China for the treatment of cognitive deficits in mild to moderate AD (
81,
82). This agent improved cognition above baseline and promoted sustained cognitive benefit through the end of a 9-month trial. Oligomannate may act through effects on the microbiome to produce both cognitive-enhancing and disease-modifying effects (
83).
The AD drug development pipeline currently has two cognitive enhancers in clinical trials in phase 1, six in phase 2, and four in phase 3 (
Figure 2). These agents exploit novel neurochemical pathways and seek to affect cognitive function either through indirect effects on cholinergic function or through effects on heretofore unaddressed neurochemical pathways.
Disease-Modifying Therapies
DMTs are being developed to prevent or delay the onset of cognitive decline in preclinical AD or to slow the progression of cognitive losses in prodromal AD or AD dementia (
24,
84). Neuropsychiatric syndromes might be expected to be affected by DMTs. Behavioral syndromes such as apathy, anxiety, and depression occur in preclinical AD (
85); neuropsychiatric syndromes emerge steadily throughout the course of the illness to more severe disorders such as agitation and psychosis (
86), and amelioration of the emergence of these symptoms can be anticipated with DMTs. Emergence analysis interrogating drug-placebo differences in the incidence of new neuropsychiatric symptoms in participants who had no or few behavioral changes at baseline is the optimal approach to assessing this type of preventive neuropsychiatric effect (
15).
Table 2 shows the National Institute on Aging–Alzheimer’s Association Common AD Research Ontology (CADRO) with classes of interventions recognized for DMT drug development in AD (
87,
88). The CADRO defines recognized disease processes in AD; these are the drug targets and related putative mechanisms of action of DMTs. The target categories include Aβ; tau protein; ApoE, lipids, and lipoprotein receptors; neurotransmitter receptors; inflammation; oxidative stress; proteostasis and proteinopathies; metabolism and bioenergetics; vascular targets; growth factors and hormones; synaptic plasticity and neuroprotection; epigenetics; neurogenesis; and “other/multi-target.”
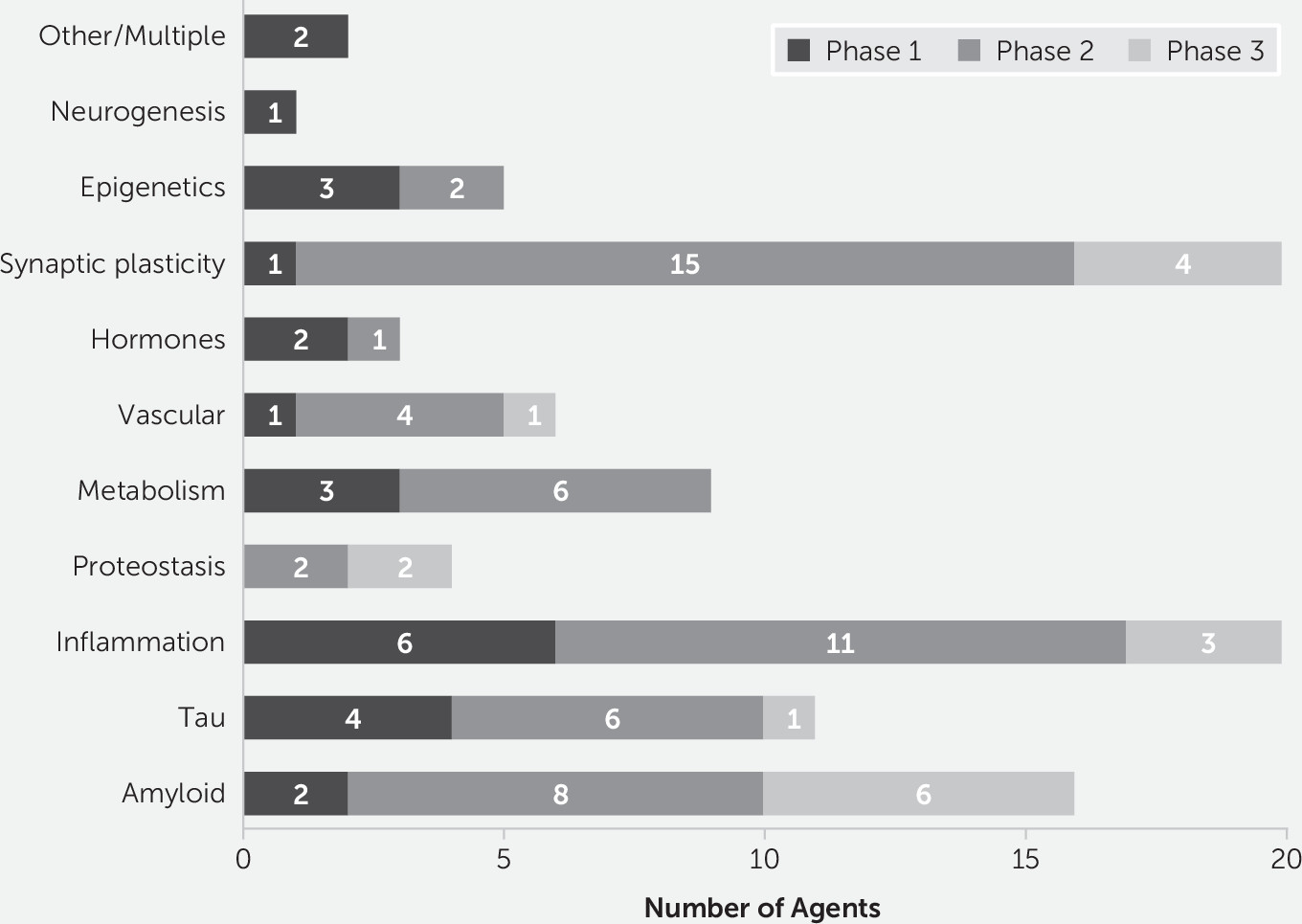
Figure 4 shows the number of agents in each phase of drug development for each target process.
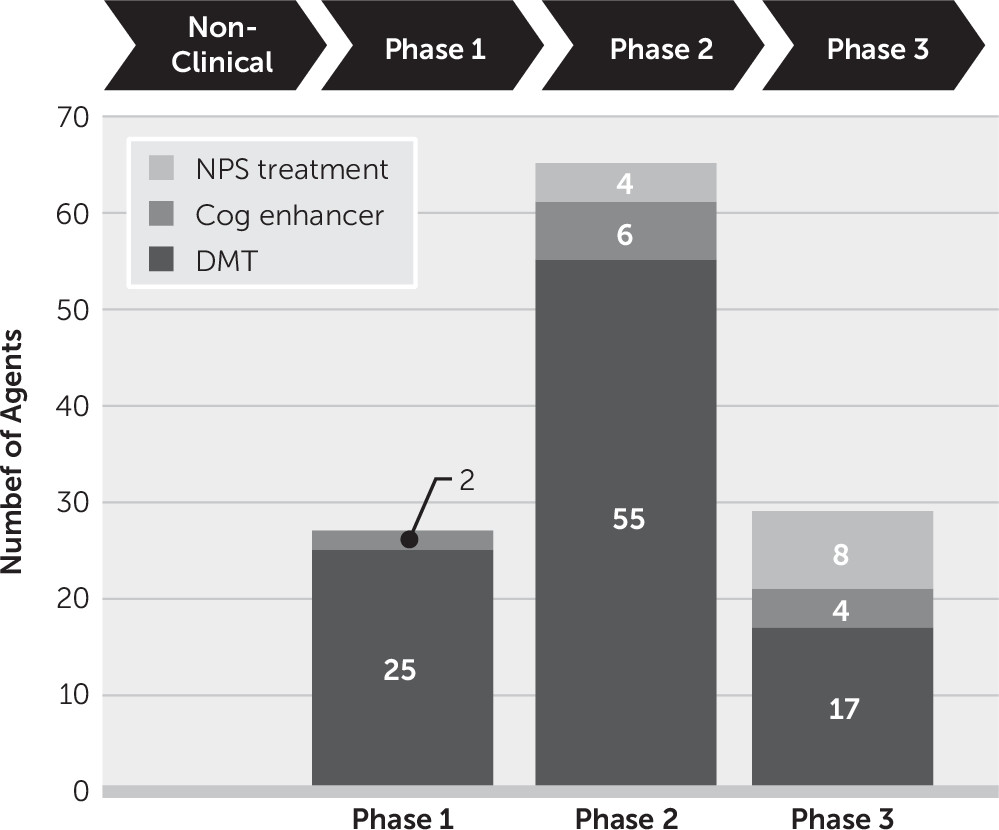
In phase 1, 93% (25 of 27) of agents in trials are DMTs (
6). Inflammation is the most common phase-1 target (six agents), followed by tau (four agents), metabolism and energetics (three agents), epigenetics (three agents), Aβ (two agents), growth factors or hormones (two agents), and one agent each for vascular targets, synaptic plasticity/neuroprotection, and neurogenesis (
Figure 3). Two agents have multiple targets.
Phase 2 has the largest number of therapies in trials compared with other phases. There are 65 drugs and biological therapies, of which 55 (85%) are DMTs (
Figures 2 and
3). Synaptic plasticity and neuroprotection are the most common targets (15 agents); inflammation (11 agents), Aβ (eight agents), metabolism and biogenergetics (six agents), tau (six agents), and vascular targets (four agents) are well represented in the phase-2 repertoire of therapies. A few sponsors are addressing proteostasis and epigenetics (two agents each) and hormonal approaches (one agent). Trials of DMTs are usually conducted in participants who are receiving stable therapy with ChE-Is, with or without memantine. Participants are randomized to the test agent or placebo, with both groups receiving the existing standard of care.
There are 29 agents being studied in phase 3; 17 are DMTs (59%) (
Figures 1 and
2). Six of these address amyloid targets, four have synaptic plasticity or neuroprotection as their target, three address inflammation, two are directed at proteostasis, and all target tau and vascular targets.
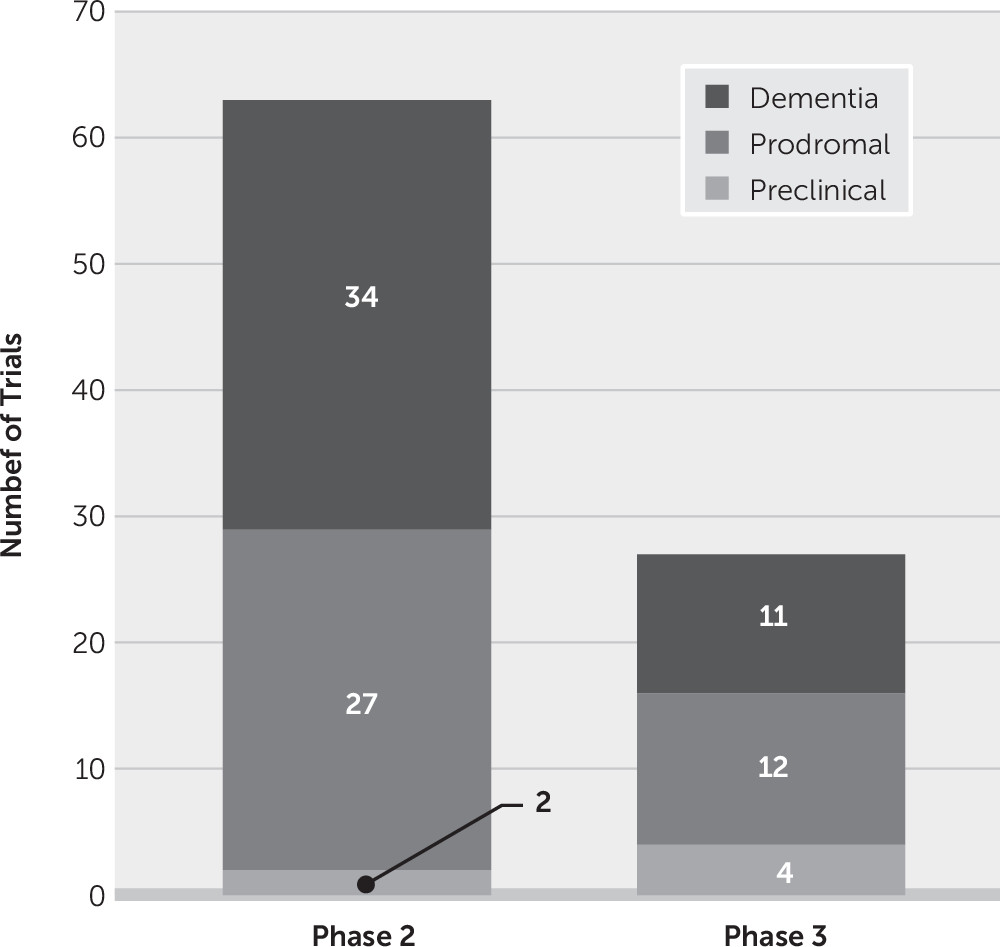
DMTs can be assessed in any phase—preclinical, prodromal, AD dementia—where the test agent may have an effect on disease progression. Cognitive-enhancing agents and psychotropic drugs must be tested in symptomatic patients in prodromal or dementia phases of AD. Of all clinical trials currently in progress, six involve preclinical populations, 39 are in patients with prodromal disease, and 45 are in patients with AD dementia (
Figure 5).
Discussion
AD drug development is progressing with current clinical trials of 121 candidate therapies. Neuropsychiatric symptoms, cognitive enhancement, and disease modification are being addressed. Prevention trials are pursued in participants who are cognitively normal and at high risk for development of symptomatic AD. There is a diversification of therapeutic targets with an emphasis on amyloid, tau, inflammation, and synaptic plasticity or neuroprotection.
Innovation in clinical trial design for assessment of treatment for neuropsychiatric symptoms is evident among recently conducted trials. The sequential parallel comparative design employed in trials of psychotropic and analgesic research was used in a development program of dextromethorphan/quinidine for the treatment of agitation in AD (
89). By rerandomizing placebo nonresponders to drug or placebo in the second stage of a two-stage trial, this design allows reduction of placebo responses that are robust in many agitation trials. A trial of pimavanserin for dementia-related psychosis used a randomized discontinuation design to demonstrate efficacy (
73). This design has the advantage of placing everyone on treatment at trial entry for 3 months before responders are randomized to continued treatment or placebo. The outcome of the trial is relapse in the placebo group compared with relapse in the active treatment group. This trial included five types of dementia, an approach that builds on accumulating evidence that psychosis is an endophenotype with shared involvement of a common brain circuitry that transcends diagnostic categories. These trial innovations expand the toolkit of available approaches to solving challenges associated with neuropsychiatric drug development.
There are no phase-1 agents for neuropsychiatric symptoms in the current AD pipeline. This reflects a multiplicity of convergent influences. First, most neuropsychiatric agents are developed for major depression, schizophrenia, anxiety, or sleep in non-AD development programs and then repositioned for AD after approval for a psychiatric disorder. Phase 1 of these agents is accomplished in programs devoted primarily to psychiatric conditions. Second, the specific biology of neuropsychiatric symptoms in AD is unknown, and there are few avenues for initiating development of psychotropics specific to the biology of AD or other neurodegenerative disorders. Third, some phase-1 trials are conducted outside the United States, are not registered on ClinicalTrials.gov, and would not be captured in our review strategy. Finally, there are too few agents entering the AD drug development pipeline for all classes of therapeutic agents. Given the 7.6-year lag between entering phase-1 trials and exiting phase-3 trials, the dearth of agents in phase 1 represents a major concern for availability of new therapies for AD in the future.
Sleep disorders in AD are underrecognized and undertreated (
90). There are a small number of sleep-related agents in the AD drug development pipeline. Suvorexant, a dual orexin antagonist, had a successful clinical trial for insomnia in AD (
20). Lemborexant, another dual orexin antagonist, is in a clinical trial for treatment of irregular sleep-wake rhythm disorder. Zolpidem and zoplicone are in a trial for insomnia in AD. These trials build on the improved understanding of the importance of sleep disorders in AD (
90).
The majority of the AD pipeline treatments are DMTs; 59% of phase-3 agents, 85% of phase-2 agents, and 93% of phase-1 agents target disease modification. Aβ protein in amyloid plaques, a variety of preplaque amyloid species, and tau protein in the form of soluble oligomers or neurofibrillary tangles are important targets in the AD DMT pipeline. Three monoclonal antibodies—aducanumab, gantenerumab, and BAN2401—have been shown to reduce brain levels of plaque amyloid and to affect CSF biomarkers indicative of neurodegeneration (
91,
92). Continuing trials of these agents will determine if they produce clinical benefit.
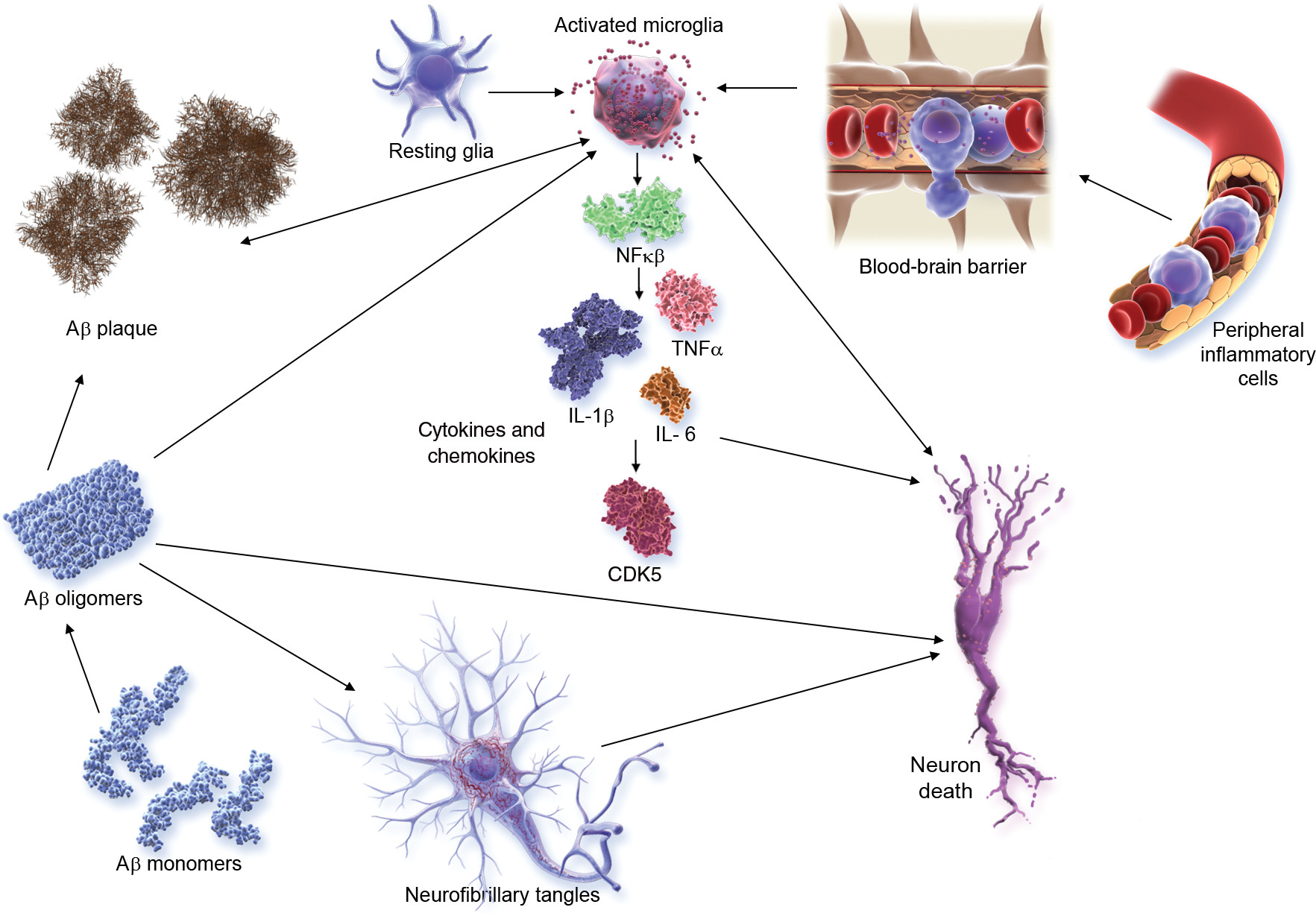
Inflammation is increasingly recognized as playing a major role in AD and other neurodegenerative disorders (
93). Microglial activation and related aspects of inflammation have emerged as important targets in the AD pipeline; agents targeting inflammatory processes are the most common approaches in both phase 1 and phase 2 (
Figure 3).
Figure 6 shows the inflammatory pathways activated in AD and targeted by the anti-inflammatory agents in trials.
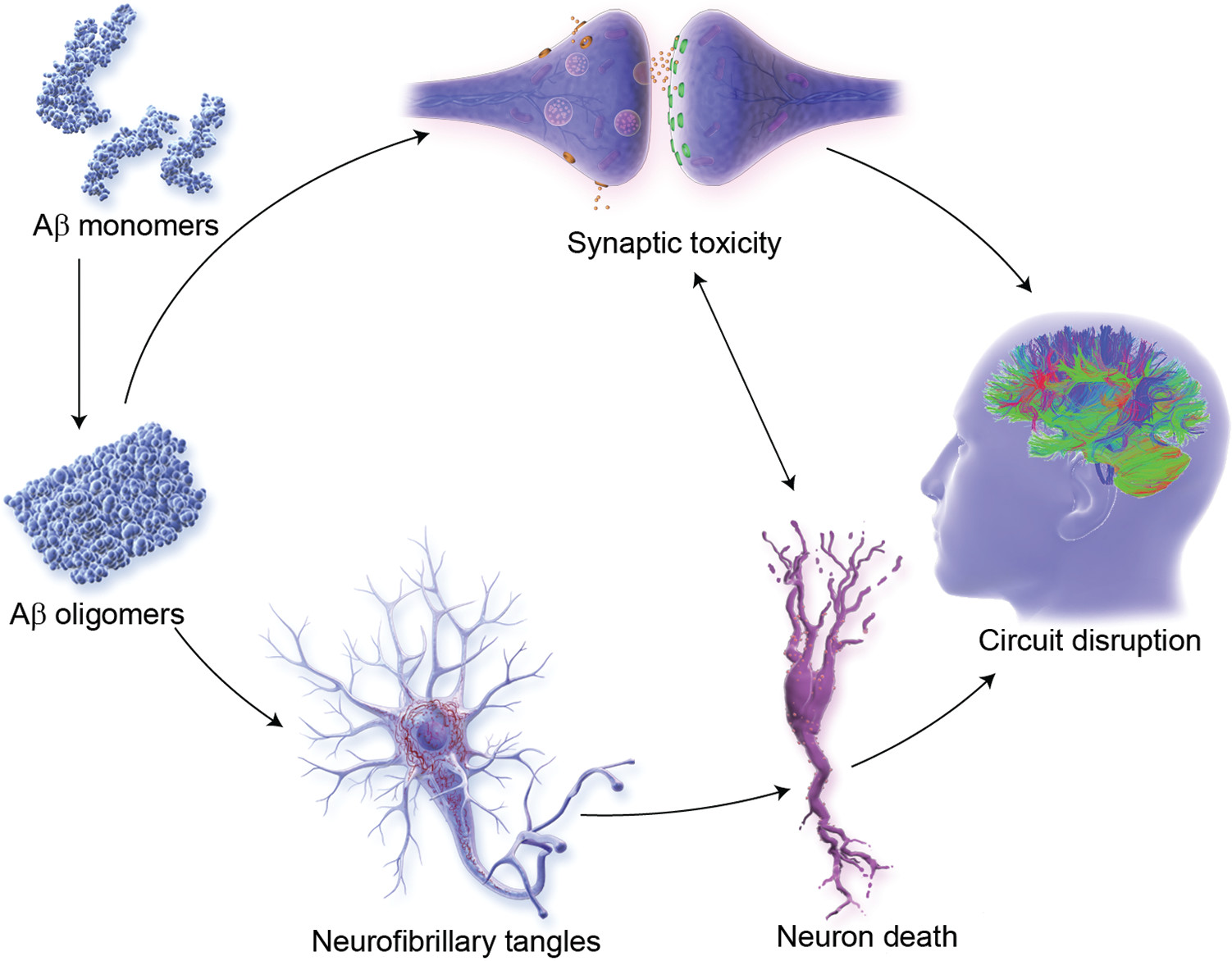
Therapies promoting synaptic plasticity and neuroprotection are well represented in phase 2, with 15 agents of this class in clinical trials (
Figures 3 and
7). These agents seek to promote synaptic plasticity or to protect synapses and neurons against Aβ or other neurotoxins (
94). Neuroprotection is the critical outcome of disease-modifying strategies (
59). These interventions promote circuit function that underly cognitive and behavioral function (
95). Success with these agents would be anticipated to have both cognitive and behavioral benefit.
The AD drug development pipeline shows the dynamic interaction between basic science and the increased understanding of the biology of AD with the development of new therapies targeting processes whose modulation may produce therapeutic benefit. This improved scientific foundation for AD drug development coupled with innovative trial designs, new biomarkers, improved clinical outcomes, and better definitions of clinical trial populations promises to accelerate delivery to new therapies to individuals with or at risk for AD.