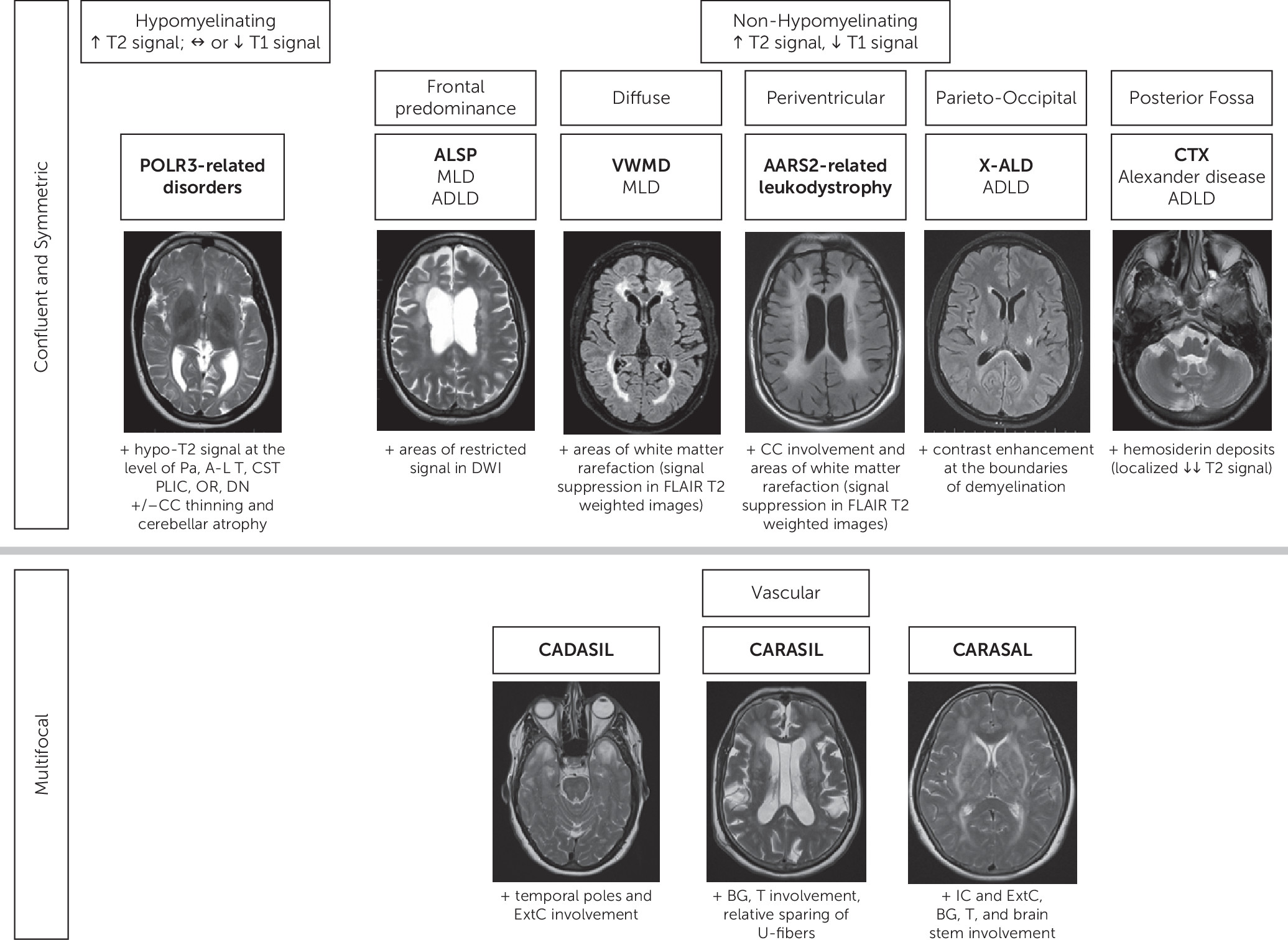
With the recent major developments in genetic and imaging techniques, an increasing number of genetic leukoencephalopathies have been identified in the adult population (
1–
5). While inherited white matter disorders are classically known to present in childhood with a dramatic clinical onset and a rapidly progressive course, affected adults show an insidious clinical presentation with a wide phenotypic spectrum, including neuropsychiatric symptoms, movement disorders, ataxia, and upper-motor neuron signs. Indeed, slowly progressive cognitive deterioration or psychiatric disturbances often dominate at presentation and can precede neurologic signs for many years. Clinicians caring for patients with various psychiatric issues should be aware of these conditions and be vigilant for clinical cues of neurologic or extraneurologic involvement indicating the need for further investigation.
Adult genetic leukoencephalopathies with psychiatric manifestations can be divided into two groups. The first group consists of adult-onset forms of hereditary leukoencephalopathies, in which insidious and often nonspecific psychiatric symptoms (e.g., mild developmental delay, learning difficulties, attention deficit hyperactivity disorder [ADHD]-like symptoms, including disturbances in sustaining attention, impulse control, and hyperactive behavior, and features of conduct/oppositional defiant disorder) can be identified earlier in life (
1,
6). Examples of such disorders are vanishing white matter disease/eIF2B-related disorders (
7), POLR3-related leukodystrophy (
8,
9), metachromatic leukodystrophy (
10), X-linked adrenoleukodystrophy (cerebral subtype) (
11), and adult-onset Alexander’s disease (
12,
13). The second group includes forms with psychiatric symptoms manifesting exclusively in adulthood. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (
14,
15), cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) (
16), cathepsin-A-related arteriopathy with strokes and leukoencephalopathy (CARASAL) (
17), adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) (
18),
AARS2-related leukoencephalopathy, adult-onset autosomal dominant leukodystrophy (ADLD) (
19), and cerebrotendinous xanthomatosis (
20) belong to this group. Often, psychiatric symptoms develop in the context of progressive cognitive decline (
1,
6).
With its high sensitivity for white matter abnormalities, MRI has a central role in the classification of leukoencephalopathies according to their patterns of involvement (
6,
21–
25). The interpretation of white matter abnormalities in association with clinical findings can orient genetic testing and allow clinicians to identify the causal gene. However, despite recent advancements in genetic techniques, and specifically the advent of next-generation sequencing (NGS), a considerable proportion (40%−70%) of adult genetic leukoencephalopathies remain without a definite molecular diagnosis (
2,
21,
26).
MRI patterns are more difficult to interpret in adult patients compared with pediatric patients due to the presence of comorbidities that can affect white matter. The diagnosis of adult-inherited white matter disorders is further complicated by unclear family history as a result of high intrafamilial clinical variability, unavailable familial DNA, and clinical and imaging features mimicking acquired disorders, such as atypical multiple sclerosis and ischemic disease (
5,
22). Consequently, adult patients with a genetic leukoencephalopathy may experience an extensive delay before receiving a definite diagnosis. This may lead to protracted inappropriate therapy and, in some cases, delayed exposure to time-sensitive treatment. Moreover, these patients and their families cannot benefit from genetic counseling and prenatal testing. Thus, it is important to define clinical and MRI pointers to accelerate the diagnosis of adults affected by genetic leukoencephalopathies.
In the present review, we provide a comprehensive overview of the adult leukoencephalopathies that can have a psychiatric presentation. We illustrate these pathologies from selected clinical cases. Additionally, we provide clinical red flags to determine when to consider genetic investigations. Finally, we adapted an MRI algorithm from those previously published to guide the interpretation of imaging findings and thus direct diagnostic investigations (
6,
21,
24).
Methods
A systematic search strategy was applied to the PubMed database. Initially, we specified no definite time period so as to obtain all cases ever reported. As a first step, we used the following search terms: (leukodystrophy) OR (leukoencephalopathy) OR (“white matter disease”) AND (genetic OR inherited OR hereditary) AND (neuropsychiatric OR psychiatric OR behavioral OR behavioral OR psychosis OR mood) AND (onset OR diagnosis OR presentation OR disturbance OR changes OR manifestations). We applied the following filters: adolescents, 13–18 years; adults, ≥19 years; and humans.
A total of 787 articles were obtained, and all were screened by title and abstract. If relevant, the entire article was read. Articles included case reports, epidemiological studies, systematic reviews, meta-analyses, and genetic studies. Only articles describing genetic, pathologic, clinical, or neuroimaging findings and algorithms for humans aged ≥16 years were included. From these articles, 16 adult genetic leukoencephalopathies were identified in which psychiatric symptoms dominate at presentation.
As a second step, we separately searched each of the 16 identified disorders in PubMed using the following search terms summarized in Table S1 in the online supplement: AND (neuropsychiatric OR psychiatric OR behavioral OR behavioral OR psychosis OR mood) AND (onset OR diagnosis OR presentation OR disturbance OR changes OR manifestations). Filters applied were as follows: adolescents, 13–18 years old; adults, ≥19 years old; and humans. Pertinent articles were selected and read in their entirety based on their titles and abstracts and whether they contained genetic, pathologic, clinical, neuroimaging, or treatment information. The present article includes only the most recent studies, published between 1992 and 2019.
Globoid cell leukodystrophy and Pelizaeus-Merzbacher disease were finally excluded because any psychiatric involvement occurred only in the advanced stages of disease and was preceded by neurological changes. Late-onset GM2 gangliosidosis was excluded because of the lack of marked white matter abnormalities in the adult form. Dentatorubral-pallidoluysian atrophy was excluded because it is a primary gray matter disease with secondary white matter involvement. Therefore, this review describes 12 adult genetic leukoencephalopathies.
Results
Adult-Onset Leukoencephalopathy With ALSP
Clinical vignette 1.
This case was previously reported by La Piana et al. (
27).
A 45-year-old patient of Croatian origin was examined for psychiatric symptoms accompanied by a recent deterioration in neurological status. At 39 years of age, she presented progressive anxiety, apathy, and feelings of sadness. These were attributed to severe depression, and she was treated initially with selective serotonin reuptake inhibitors and then with risperidone. Medications had very limited benefit. Memory problems and executive dysfunction later complicated the picture. She eventually had to take a leave of absence from her work due to increasing difficulties with organization and coping with anxiety.
Before the age of 39, the patient had never previously exhibited mood or cognitive disturbances and had a negative neurological history. Family history, however, was significant for bipolar disorder in her mother and psychosis in her older sister.
At the age of 44, neurological signs appeared as progressive shuffling gait, rigidity, and difficulty finding words. She also demonstrated perseverative behaviors. In this context, she was referred to our team for an opinion and workup. On clinical examination at the age of 45, the patient exhibited severe anxiety and emotional lability and provided limited insight into her symptoms. The neurological examination documented saccadic pursuits, cogwheel rigidity, brisk reflexes, shuffling gait, and impossible tandem gait. She scored 22/30 on the Mini-Mental State Examination and 18/30 on the Montreal Cognitive Assessment. Brain MRI documented confluent, bilateral white matter lesions, predominant in the frontal region with mild asymmetry (R>L) (
Figure 1).
A brain biopsy showed enlarged irregular axons with spheroids. The genetic confirmation was provided by the CSF1R gene sequencing (NM_005211.3), which revealed a novel heterozygous variant (c.2350G>A; p.V784M). The patient was therefore diagnosed with hereditary diffuse leukoencephalopathy with spheroids (HDLS). We identified the same pathogenic mutation in the patient’s mother, therefore demonstrating a high intrafamilial phenotype variability.
ALSP is one of the most frequent adult leukodystrophies; it comprises two clinically and genetically similar entities, known as HDLS and pigmentary orthochromatic leukodystrophy (
28–
30). Disease onset occurs at a mean age of 42 years, usually earlier in women than men (40 years old versus 47 years old), and is characterized by slowly progressive cognitive dysfunction and behavioral and personality changes (
18,
28). Neuropsychiatric manifestations represent the initial symptom in 44% of patients with ALSP (
31). Following, or occurring simultaneously, are motor impairments, including gait difficulties and pyramidal signs. Parkinsonism may develop (
31). Disease course is very rapid with an average of 3.9 years from onset of symptoms to loss of capacities and 6.8 years from onset to death (
31).
Typical MRI findings include frontal and parietal predominance of white matter abnormalities, often asymmetric, with secondary dilation of the lateral ventricles and thinning of the corpus callosum (
Figure 1) (
32). Areas of diffusion-weighted imaging (DWI) restricted diffusion can be documented (
33). Calcifications were detected in the abnormal white matter in 54% of cases (
28,
33).
ALSP displays an autosomal dominant pattern of inheritance with incomplete penetrance. It was also reported as sporadic. It is caused by mutations in
CSF1R, encoding the colony stimulating factor 1 receptor. This receptor ensures the proliferation and survival of mononuclear phagocytic cells, including microglia of the central nervous system (
28). Clinical features of neuropsychiatric and motor impairments and MRI findings suggest the diagnosis, which is confirmed by
CSF1R sequencing (
29).
AARS2-Related Leukoencephalopathy
Mutations in the
AARS2 gene are associated with a progressive leukoencephalopathy, which may present with neuropsychiatric, cognitive and extrapyramidal features and wide phenotypic variability (
34). Twenty cases of
AARS2-related leukoencephalopathy have been reported, in which psychiatric symptoms usually precede neurological motor symptoms. Disease onset occurs at a mean age of 27.3 years, with subsequent development of neurological symptoms at a mean age of 33 years (
35). Patients demonstrate rapid deterioration and become bedridden in the following 3–5 years on average (
36,
37). Common psychiatric complaints include changes in personality, mood fluctuation, difficulties sustaining attention and planning, memory problems, psychosis, and behavior disinhibition. Additionally, cognitive decline with features of frontal lobe dysfunction, pyramidal signs, and cerebellar ataxia are commonly observed. All reported females had primary or secondary amenorrhea resulting from ovarian failure (
37–
39).
Brain MRI documents white matter abnormalities predominantly in the frontal and parietal periventricular regions, with involvement of the corpus callosum and corticospinal tracts. White matter rarefaction is often observed in the peritrigonal regions (
Figure 1). Patchy areas of restricted diffusion are sometimes documented in diffusion-weighted imaging (
35,
37,
39)
The disorder is inherited in an autosomal recessive pattern and caused by mutations in AARS2, which encodes mitochondrial alanyl-tRNA synthetase 2.
In the context of suggestive clinical and neuroradiological findings, the identification of pathogenic mutations in
AARS2 sequencing allows the diagnosis. For the overlapping MRI and clinical picture, screening for
AARS2 mutations is recommended in patients with suspected ALSP who are
CSF1R-negative (
35,
39).
Adult-Onset Alexander’s Disease (AOAD)
AOAD accounts for approximately 22% of cases of Alexander’s disease. It manifests as a large phenotypic spectrum characterized by brainstem and cerebellar dysfunction evident as bulbar or pseudo-bulbar signs, gait ataxia, and spasticity (
40). Patients may also display neurobehavioral changes reflecting behavioral-variant frontotemporal dementia without its specific MRI findings (
13). Attention deficit, executive dysfunction, expressive deficits, and memory loss characterize the neuropsychological symptomatology. In addition, patients may show a constricted affect, impulsivity, and symptoms of depression, which progress to social and occupational impairment in advanced disease (
13).
Typical brain MRI findings in adult patients are characterized by a periventricular band of white matter signal changes and infratentorial tadpole-like images representing medulla and upper-cervical cord atrophy. Signal changes in the hilum of the dentate nuclei are common in AOAD. Abnormalities can enhance postcontrast injection (
12,
40,
41).
AOAD is an autosomal dominant disease caused by mutations in the
GFAP gene that encodes the gliofibrillary acid protein, the main intermediate filament of differentiated astrocytes (
40). As opposed to what is seen in pediatric populations, where the majority of cases are sporadic, AOAD patients may have an affected parent (
42,
43).
Adult-Onset ADLD
ADLD is a rare neurodegenerative disorder with onset in the fourth or fifth decade of life. The first manifestations include autonomic dysfunction, progressive pyramidal signs, and ataxia, usually followed by slow cognitive decline and depressive symptoms (
44). However, Jain et al. (
45) reported a 30-year-old patient with an 8-year history of rapid-cycling behavioral disorder with mood disturbances, logorrhea, sexual promiscuity, and flight of ideas, followed only 4 years later by neurological manifestations. The median life expectancy is 18 years after disease onset (
46).
Brain MRI documents predominantly frontal and parietal white matter abnormalities that can extend to the occipital region, with sparing of the periventricular regions and U-fibers. The involvement of the corpus callosum, cerebral peduncles, and pontine middle cerebellar peduncles is typical (
19,
47). MRI changes often precede the clinical manifestations by many years (
46). The spinal cord can be affected by demyelination and atrophy (
48). ADLD is caused by either heterozygous duplication on chromosome 5q23 that leads to lamin B1 (LMNB1) overexpression in brain tissue or deletions upstream of the gene (
32,
46,
49,
50).
ADLD should be considered in any patient with psychiatric symptoms and autonomic dysfunction. Evidence of white matter abnormalities further supports the diagnosis, which is confirmed by the identification of pathogenic duplications or, more rarely, upstream deletions involving the
LMNB1 gene (
51,
52).
POLR3-Related Leukodystrophy
Clinical vignette 2.
A 24-year-old female was referred to our neurogenetics clinic following the incidental finding of diffuse leukoencephalopathy disclosed on an MRI of the sella turcica conducted in the context of primary amenorrhea. She is the firstborn of nonconsanguineous French-Canadian parents with a negative family history for neurologic or psychiatric illness. Since primary school, she had experienced very mild learning difficulties, which were not formally assessed until 3–4 years prior to presentation. She had more recently complained of worsening attention and concentration and episodes of lack of judgement that caused her to drop out of school and to be dismissed by various employers. In relation to the slowly progressive symptoms, she developed increasing anxiety. At age 15, she had consulted an endocrinologist for primary amenorrhea, which remained without diagnosis. She reported no other medical complaints except for severe myopia. On examination, her mood and cognition seemed within normal limits. The neurological examination was normal except for overall increased deep tendon reflexes and clonus.
Brain MRI documented bilateral and symmetric white matter abnormalities, hypomyelinating in origin (isointense on T
1-weighted images and hyperintense on T
2-weighted images), with inhomogeneous signal intensity. They were associated with mild T
2-hypointense signal at the level of the pallida nuclei and anterolateral nucleus of the thalami (
Figure 1). We did not observe T
2 hypointensities at the level of the dentate nuclei or the posterior limb of the internal capsule.
Genetic testing revealed two previously unreported predicted pathogenic variants of the POLR3A gene (c.2080C>T, p.Arg694Cys; c.3020C>T, p.Thr1007Ile). Each parent was a carrier of one variant. The patient was therefore diagnosed with POLR3-related leukodystrophy, which was compatible with the imaging results, neuropsychological impairment, severe myopia, and primary amenorrhea.
POLR3-related leukodystrophy encompasses five overlapping clinical syndromes previously described separately as 4H syndrome (hypomyelination, hypodontia, hypogonadotropic hypogonadism), ADDH (ataxia, delayed dentition, and hypomyelination), TACH (tremor-ataxia with central hypomyelination), LO (leukodystrophy with oligodontia), and HCAHC (hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum) (
8,
53). POLR3-related leukodystrophy is progressive and life limiting, with late-onset cases surviving until their fourth or fifth decade of life (
8).
Neurological manifestations mainly revolve around cerebellar dysfunction and present as gait ataxia, dysarthria, dysmetria, tremor, and abnormal eye movement. There may also be extrapyramidal signs such as dystonia (
54,
55). In late-onset POLR3-related leukodystrophy, which represents approximately 10% of cases, learning difficulties may be the presenting symptom. It may also be associated with behavioral disturbances (
8).
Nonneurologic features include abnormal dentition (e.g., hypodontia, oligodontia, or delayed teeth eruption), endocrine abnormalities (delayed, arrested, or absent puberty and/or short stature), and severe myopia and often suggest the diagnosis (
8,
56).
The most common brain MRI pattern is characterized by diffuse hypomyelination with relative T
2-weighted hypointensity of the pallida, anterolateral nuclei of the thalami, dentate nuclei, and pyramidal tracts in the posterior limbs of the internal capsules and optic radiations (
Figure 1). However, hypomyelination is not always present, and atypical MRI patterns are described (
57,
58). Cerebellar atrophy and thinning of the corpus callosum are commonly present (
9,
59).
POLR3-related leukodystrophy is inherited in an autosomal recessive manner. Causative mutations have been identified in four distinct genes:
POLR3A,
POLR3B, POLR1C and
POLR3K (
8,
60–
63).
POLR3A variants are related to a later disease onset, compared with
POLR3B variants, but a more rapid disease progression (
9). However, a common
POLR3B pathogenic variant (c.1568T >A [p.Val523Glu]) can be associated with either asymptomatic or mildly symptomatic disease with onset in early adulthood (
9,
64,
65).
Diagnosis should be suspected in the presence of suggestive symptoms and diffuse hypomyelination accompanied by T
2 hypointensity of the anterolateral nuclei of the thalami and/or the pallida (
59). It is confirmed by the presence of biallelic pathogenic variants on one of the four genes identified to date.
Vanishing White Matter Disease/eIF2B-Related Disorders (VWMD)
Clinical vignette 3.
This case was previously reported in Accogli et al. (
66).
A 41-year-old male patient was referred to the neurogenetics clinic for suspected genetic leukoencephalopathy discovered incidentally following a motor vehicle accident. He is the product of a consanguineous relationship between two French-Canadian first cousins. The patient’s family history was positive for psychiatric illness (suicide in the context of drug addiction in two maternal uncles and schizophrenia in the patient’s grandmother). His initial symptoms began in late childhood and manifested as severe behavioral problems marked by aggressiveness and defiance. He had a very short temper, got into violent fights at school, and exhibited disobedience with all authoritative figures. He also sexually abused a family member. At 16 years old, he suffered his first episode of depression and attempted suicide. Eventually, his behavior disturbances caused him to drop out of school. He then worked as a truck driver until the age of 40.
On further assessment of past neurological symptoms, the patient had been experiencing transient urinary incontinence since age 16. At age 34, he started noticing difficulty with concentration and short-term memory. A few years later, on hospitalization for another suicide attempt, the family noticed mild gait instability for the first time.
At age 40, he was admitted to the hospital for injuries sustained in a truck accident. Following the accident, he encountered cognitive difficulties, progressive motor impairment, and dizziness that affected his gait and led to frequent falls. A head CT scan revealed diffuse and symmetric white matter abnormalities, prompting an MRI study.
Brain MRI showed diffuse and symmetric white matter involvement with relative sparing of the U-fibers, the external rim of the corpus callosum, the internal capsule, and the cerebellum. Areas of focal white matter rarefaction were documented at the level of the frontal horns of the lateral ventricles (
Figure 1).
On clinical assessment performed at the age of 41, the patient was being treated with venlafaxine, with only partial benefit. He was oriented and cooperative. His mood seemed depressed. He had only partial recollection of major life events, from which he showed emotional detachment. The neurological examination was significant for mildly increased tone overall, brisk reflexes in the lower limbs, and mild dysdiadochokinesia. His gait was slightly ataxic. An NGS panel for leukodystrophies revealed the homozygous mutation c.260C.T (p.Ala87Val) in EIF2B3 (NM_020365.3), and the patient was therefore diagnosed with VWMD.
VWMD is caused by pathogenic mutations in one of the five
EIF2B1–5 genes and was initially described in children (
67,
68). However, it is now well established that VWMD can also present in late juvenile, adolescent, and adult ages. These latter forms correspond to a more slowly progressive course with lower mortality. Stress is a trigger of disease in 53% of cases and/or an aggravating factor leading to rapid neuropsychiatric deterioration in 38% of patients with disease onset after 4 years of life (
7).
The adult-onset form represents more than 15% of VWMD diagnoses (
69). Presentation is characterized by cognitive or psychiatric manifestations (depression and psychosis) in 11%−16% of cases. Neurological symptoms follow within a few years (
66,
70). In females, signs of ovarian failure (amenorrhea, irregular menses, and infertility) are described in 86% of cases (
7).
Diffuse and symmetric white matter abnormalities are typically observed in affected patients, including the corpus callosum. A hallmark of the disease is the progressive white matter rarefaction visible on fluid-attenuated inversion recovery T
2-weighted and proton density sequences (
Figure 1) (
24,
69,
71).
VWMD is an autosomal recessive disorder caused by biallelic mutations in any of the
EIF2B1-5 genes, which encode for the five subunits of eiF2B. The eiF2B protein complex is crucial in the regulation of protein synthesis, especially during stress (
7,
72).
Recognizing VWMD in early stages is essential in order to take preventative measures to avoid stress-provoking episodes, such as head trauma and fever, which are the main predictors of disease course (
7). It should be considered in patients with identifiable triggers leading to a rapid decline and compatible MRI findings. VWMD is confirmed by molecular genetic testing.
Inborn Errors of Metabolism
Inborn errors of metabolism are disorders that are potentially treatable; therefore, promptly detecting their psychiatric presentation becomes a crucial concern for clinicians.
Cerebrotendinous Xanthomatosis (CTX)
CTX is a rare autosomal recessive lipid storage disorder. Heterogeneous clinical presentation often leads to a diagnostic delay of up to 25 years (
73). Although psychiatric symptoms as the initial manifestation have been reported sporadically, they can be the first sign prompting the patient to seek medical attention. It is thus important to investigate the patient’s past medical history to search for common systemic manifestations such as chronic diarrhea, juvenile bilateral cataracts, tendon xanthomas, premature atherosclerosis, pulmonary dysfunction, and osteoporosis, as well as neurological signs, including psychomotor retardation, cognitive dysfunction, cerebellar ataxia, and epilepsy. These features can orient toward the diagnosis (
73). Approximately 50% of patients have a psychiatric presentation, including mood disorders (anxiety and depression as well as bipolar disorder), delusional disorder, and psychosis, with occasional reports of catatonia (
20,
73,
74). In most cases, these are refractory to pharmacotherapy and psychotherapy (
73).
MRI findings are distinctive and characterized by T
2-weighted hyperintensities in the cerebellar hemispheres and dentate nuclei, brainstem, and periventricular white matter. Calcium and hemosiderin deposits around the dentate nuclei often orient the diagnosis (
Figure 1). Spinal cord involvement is possible, with lateral and dorsal column involvement on T
2-weighted images (
75).
CTX is caused by recessive mutations in the
CYP27A1 gene, leading to the dysfunctional conversion of cholesterol into bile acids. Large deposits of the synthetic pathway by-products of bile acids accumulate in many organs and in myelin, leading to progressive demyelination (
74).
CTX should be suspected in a patient with suggestive clinical features and MRI findings. Elevated CSF or plasma cholestanol levels support the diagnosis, which is confirmed by evidence of mutations on
CYP27A1 sequencing (
74).
The treatment consists of chenodeoxycholic acid, which can be combined with a statin for an increased antiatherosclerosis effect. Early diagnosis and treatment can prevent disease progression, alleviate symptoms, and improve prognosis (
73).
Metachromatic Leukodystrophy
Clinical vignette 4.
A 23-year-old male was investigated for behavioral disturbances and increasing school difficulties. He is the firstborn child of nonrelated French-Canadian parents, and his family history is negative for neurological or psychiatric disorders.
The patient’s symptoms started around the age of 15, when the parents noticed a decline in his school performance, attention span, and interest for learning. This contrasted with the patient’s prior academic achievements. In this context, he underwent a first evaluation with the school psychologist and received the diagnosis of ADHD.
Over time, he developed behavioral problems with increasing aggressiveness. Meanwhile, he was not able to keep his job as a grocery stocker due to lack of autonomy in the tasks. This behavior, together with the ADHD symptoms (which were refractory to atomoxetine, risperidone, and lisdexamfetamine), prompted a psychiatric evaluation and a full neuropsychological assessment. The latter identified mild cognitive deficit with considerable difficulties in information processing, concentration, and executive function.
Following a severe, aggressive outburst, a brain MRI was performed to exclude the presence of an underlying organic condition. The examination documented bilateral and symmetric white matter abnormalities in the frontal and parieto-temporal regions (
Figure 1). The white matter had a questionable “striated” appearance. Electromyography showed severe sensorimotor demyelinating polyneuropathy.
On clinical examination, at the age of 23, the patient was cooperative and understood the tasks of the examination. He impulsively asked questions that were sometimes unrelated. The neurological evaluation showed dysarthria, dysmetria, absent reflexes in the lower limbs, and reduced vibration sensation distally in the lower extremities. His gait was normal, but he had difficulty with the tandem gait.
An NGS panel for genes related to hereditary leukoencephalopathies demonstrated two variants in the ARSA gene (NM_000487) (c.542T>G; p.Ile181Ser and c.495_501del; Pro156Leu fs*32). Blood arylsulfatase levels were below normal. These data prompted the diagnosis of metachromatic leukodystrophy (MLD).
MLD is a severe inherited neurometabolic disease that can present during infancy, childhood, adolescence, or adulthood. Both juvenile and adult MLD most commonly manifest with cognitive and psychiatric features typical of frontal lobe dysfunction, usually behavioral disturbances and psychosis (
76,
77). Peripheral neuropathy, pyramidal signs, and ataxia often follow gradually. Subsequent symptoms develop over the following 5–35 years and include spastic tetraparesis, incontinence, bulbar palsy, dementia, and seizures (
78). Up to 75% of patients have gallbladder involvement (e.g., thickened gallbladder wall, polyps, and gallbladder cancer) and may thus present with abdominal pain (
79).
Psychosis in MLD is characterized by disorganized thought content, complex auditory hallucinations, and bizarre delusions. As a result of the presence of psychotic disorder in approximately 50% of cases and the initial absence of neurological findings, many MLD patients are misdiagnosed with schizophrenia and treated accordingly without benefit (
77,
80).
In the juvenile and adult forms, confluent white matter abnormalities are observed in the frontal and parietal regions, with involvement of the corpus callosum and relative sparing of the U-fibers (
Figure 1). Sparing of the perivascular myelin produces a “tigroid” pattern (
81).
MLD is inherited in an autosomal recessive pattern with incomplete penetrance. It is caused by biallelic mutations in the
ARSA gene, which encodes for the lysosomal enzyme arylsulfatase A. The enzymatic deficiency leads to accumulation of sulfatides in the central and peripheral nervous systems, precipitating demyelination (
78). MLD is suggested by low
ARSA activity in blood leukocytes and elevated urine sulfatides and confirmed by
ARSA gene sequencing (
81).
Hematopoietic stem cell transplantation (HSCT) has been shown to provide benefit for both juvenile and adult MLD if considered early in the disease or in the presymptomatic stage. HSCT is not recommended for the late infantile form of the disorder (
10,
78).
X-Linked Adrenoleukodystrophy
X-linked adrenoleukodystrophy (ALD) is a rare inborn error of metabolism with different phenotypes: the childhood cerebral form, adremyeloneuropathy, and the “Addison disease only” phenotype are the most common. Symptomatic female carriers present progressive spastic paraparesis and sphincteric control problems (
82). The adult-onset cerebral ALD form is one of the less frequent yet relatively rapidly progressive presentations and manifests as any combination of neuropsychiatric, adrenal, and gonadal symptoms. It occurs most commonly in the second or third decade of life (
83,
84). Initial symptoms are of a psychiatric nature and include signs of mania or psychosis. Signs of myelopathy or adrenal insufficiency and testicular dysfunction reported as a history of sexual dysfunction may also be present before onset of psychiatric disease and should orient the diagnosis (
84,
85). Further evidence of the underlying metabolic disorder includes drug resistance to neuroleptic and anticholinergic treatment with abnormal or increased side effects (
83).
Brain MRI typically documents confluent and symmetric white matter abnormalities in the parieto-occipital and temporal regions, with involvement of the splenium of the corpus callosum and cerebellum (
Figure 1). Contrast enhancement at the borders of the demyelinating lesions is a hallmark of ALD. Visual and auditory pathways are affected in up to 75% of patients (
82,
85).
ALD is an X-linked disorder caused by mutations in the
ABCD1 gene, which impairs peroxisomal fatty acid beta oxidation, leading to accumulation of very long-chain fatty acids (VLCFA) in the nervous system, the adrenal cortex, and the gonads (
85). The metabolic diagnostic test is measurement of VLCFA in the serum, and a definitive diagnostic test is the detection of a hemizygous
ABCD1 mutation. Prompt genetic counseling allows for early identification of other affected males in the family of patients that may benefit from HSCT.
Treatment with allogeneic HSCT provides the most benefit in young patients in early stages of the disease before the onset of neurological symptoms. The presence of MRI abnormalities, especially contrast enhancement, may determine the eligibility for HSCT, but the criteria for HSCT in adults are still to be defined (
83,
86).
Small-Vessel Diseases
Cadasil
CADASIL is an inherited small-artery disease clinically characterized by a heterogenous combination of migraines with aura, ischemic events, cognitive decline, and neuropsychiatric disorders. Migraines usually represent the first clinical manifestation, with onset in the second or third decade of life, while the full clinical spectrum can develop over 10–40 years (
14). Psychiatric symptoms are observed in 48%–73.9% of cases at any stage of the disease (
15,
87,
88). Close to 10% of cases involve psychiatric features as the sole symptom at onset, such as mood disturbances associated with adjustment disorders, unipolar major depression, and bipolar disorder, as well as psychotic disorders (
87,
89). Depression is the most commonly reported disturbance, followed by bipolar disorder, and somatic symptoms associated with anxiety are frequently reported in patients with depression and CADASIL (
88).
MRI findings include multifocal white matter T
2-weighted hyperintensities in the deep white matter with accompanied lacunar infarcts. The involvement of the external capsules and poles of the temporal lobes is highly suggestive of the disease (
Figure 1) (
87).
CADASIL is an autosomal dominant disorder caused by mutations in the
NOTCH3 gene, inducing thickening of the arterial wall and degeneration of smooth muscle and endothelial cells (
15). The majority of the mutations involve a cysteine residue in the epidermal growth factor-like repeats.
Diagnosis is confirmed by
NOTCH3 sequencing. The identification of granular osmiophilic material in the skin biopsy of patients is 100% concordant with the presence of pathogenic mutations (
90).
Carasil
Like CADASIL, CARASIL is a rare inherited disease particularly common in patients of Asian descent in which recurrent ischemic events lead to stepwise deterioration of brain functions with cognitive decline. Alopecia and lumbago/spondylosis deformans are cardinal clinical features (
91).
Memory dysfunction is an early finding in patients with CARASIL, usually by the third to fourth decade of life (
90), and is associated with disorientation, behavioral disturbances, compulsive behavior, and personality changes, including irritability and emotional lability (
92). Unlike in CADASIL and CARASAL, depression is uncommon (
93).
Brain MRI shows T
2-weighted hyperintensities in periventricular, frontal, and temporal white matter, occasionally involving the external capsule and pons in the early stages and middle cerebellar peduncles in late stages of the disease, with relative sparing of the U-fibers (
Figure 1). Multiple lacunar infarcts and microbleeds are observed in the basal ganglia and thalami (
90,
91).
CARASIL is caused by recessive mutations in the
HTRA1 gene, encoding for the HTRA1 serine protease (
90). Recently, a dominant variant of the disease, caused by heterozygous
HTRA1 mutations, has been reported (
92,
94–
97).
CARASIL should be considered in the presence of leukoencephalopathy with ischemic changes, especially when associated with alopecia or lower back pain and when the patient is of Asian ethnicity (
93). Genetic testing is the gold standard for diagnosis of CARASIL.
Carasal
CARASAL is a rare condition that to our knowledge is described in only 19 cases to date. Migraine is the most common manifestation, followed by stroke, cognitive impairment, movement disorder, dysphagia, gait disturbance, and depression (
17). In one patient, the initial symptoms were mild behavioral changes and disinhibition (
2). Onset of symptoms occurs between the third and fourth decade of life. Given the small number of reported cases, the prognosis has not yet been determined, but life expectancy is not significantly reduced (
17).
Brain MRI demonstrates frontoparietal periventricular and deep white matter T
2-weighted hyperintensities sparing the U-fibers, initially multifocal and then secondary confluent (
Figure 1). The thalamus, basal ganglia, and brainstem, especially around red nuclei, are also involved. Infarcts, microbleeds, and hemorrhages become evident in older patients (
17).
CARASAL is an autosomal dominant disorder caused by a pathogenic mutation in the
CTSA gene (
2,
98,
99). It should be suspected in patients with recurrent ischemic events and leukoencephalopathy. Definitive diagnosis is obtained by identification of the causative mutation through
CTSA sequencing (
17).
Genetic Leukoencephalopathy of Unknown Origin
Approximately 40%−70% of adult patients with leukoencephalopathy of suspected genetic origin remain without definite diagnosis despite extensive testing (
2,
21,
26). Hence, undiagnosed leukoencephalopathies in adults have become an emerging challenge in clinical neurosciences. A significant percentage of these cases will have early psychiatric presentations.
Clinical vignette 5.
A 53-year-old female presented with a 5-year history of progressive cognitive decline. She is the only child of a French-Canadian mother and an African-American father. Her family history is unremarkable, although her father died at age 57 of an unknown cause.
She was born preterm at 32 weeks and was small for gestational age, requiring hospitalization for several weeks. Afterward, she reportedly had a normal psychomotor development. She completed high school and had worked as a legal secretary until the onset of her illness.
At age 48, her first symptoms manifested as memory and cognitive difficulties. This led to multiple employment terminations and subsequent financial and emotional difficulties. Two years later, she experienced severe mood changes that led to a suicide attempt. She was diagnosed with major depression, but associated findings of tremor and balance problems prompted further investigations. A first MRI demonstrated multifocal and confluent white matter abnormalities, suggesting multiple sclerosis.
Following interferon treatment, she progressed with further motor and cognitive deterioration. In the following months, she developed urinary incontinence. A second MRI documented severely confluent and symmetric cerebral and cerebellar leukoencephalopathy and moderate cerebral atrophy.
On physical examination at the age of 53, the patient exhibited limited understanding of the tasks. She could answer questions but had only partial recollection of life events. Neurological examination documented saccadic visual pursuits, hypotonia, hyperreflexia, dysmetria, and dysdiadochokinesia. The patient also displayed truncal and gait ataxia.
Extensive genetic testing, including EIF2B1–5 gene sequencing, NGS panel for mitochondrial disorder, and NGS panel for known leukoencephalopathies (203 genes plus mitochondrial DNA), was performed to rule out a genetic white matter disorder, and no pathogenic variants were detected. At present, the patient remains without a definite molecular diagnosis; a whole-exome sequencing is planned. In this patient’s case, whole-exome sequencing will be useful to identify potentially causal variants in new genes responsible for white matter disorders or in genes associated with genetic disorders not associated yet with white matter abnormalities.
Discussion
Abnormalities in white matter have been consistently reported in association with psychiatric disorders (
100–
102). The increasing number of identified adult genetic leukoencephalopathies along with their genetic and clinical heterogeneity and overlapping radiological phenotypes pose a diagnostic challenge for clinicians. We believe that this challenge may be even more difficult in patients presenting with a psychiatric onset, because their symptoms are most often misattributed to a primary psychopathology (
73,
83,
85).
Encountered diagnostic difficulties include lack of awareness of these disorders, unclear family history, and the undifferentiability between a primary psychiatric condition and the symptom of a leukoencephalopathy. Nevertheless, clinicians need to provide patients with leukoencephalopathy with a definite diagnosis as early as possible in order to refer them to a specialized center for these rare conditions, provide genetic and family counseling, and offer targeted treatments whenever these are available.
Some reviews have proposed clinical and MRI guidelines for timely recognition of adulthood genetic leukoencephalopathies in a routine clinical setting (
1,
3,
21,
103). However, none have specifically addressed affected patients with a psychiatric onset of disease. We therefore provide the following guidelines to facilitate the identification of these disorders.
Clinical Features and Elements of Family History That May Suggest an Inherited White Matter Etiology
Atypical response to treatment.
Further assessment for inherited leukoencephalopathies should be considered in a psychiatric patient when there is refractoriness to standard pharmacological treatment or when there are unusual side effects (
83,
85). It is widely known that drug refractoriness is common in psychiatry; depression has nonresponsiveness rates of 19%–34% (
104), and 10%−30% of patients with schizophrenia are considered to have treatment-resistant symptoms (
105,
106). The relatively high prevalence of such disorders compared with genetic leukoencephalopathies may lead some to consider that the cost-benefit ratio does not warrant the pursuit of further investigations. However, psychiatrists should consider performing a brain MRI to exclude genetic leukoencephalopathy in patients with atypical response to treatment because of the importance of prompt diagnosis and because specific treatments may be available for some metabolic forms (MLD, ALD, and CTX) (
10,
73,
78,
83). Ongoing studies show a slowing of symptom progression or reduction in adult patients affected by a lysosomal disorder, which have maximum benefit in early stages of the disease (
107). In addition, genetic leukoencephalopathies require specific management that includes supportive therapies as the disease progresses.
To help clinicians decide whether to request a brain MRI, drug resistance should be considered important. If one or more of the features listed below are also present, then neuroimaging is highly recommended.
Neurological and systemic features.
The presence of neurological symptoms in a psychiatric patient should prompt the clinician to further investigate with MRI. The more common presenting features are pyramidal signs; ataxia or other cerebellar features; bladder, bowel, or sexual dysfunction; peripheral neuropathy; and cognitive deficits. These features may occur simultaneously with the psychiatric manifestations or may develop later. In some rapidly progressive disorders such as ALSP and
AARS2-related leukoencephalopathy, the neurological symptoms may appear months after the psychiatric presentation. However, in more slowly evolving diseases, the neurological involvement may present much later, even years after the first psychiatric symptoms. In other cases, subtle and insidious neurological symptoms can precede the psychiatric presentation (
66). Developmental delay, ADHD, or lifelong learning disabilities in the pediatric history may be the presenting symptoms for patients with POLR3-related leukodystrophy, as described in the clinical vignette. Ideally, neurological examination in psychiatric patients should be performed as part of their initial evaluation and repeated over time if other warning signs are present or appear (e.g., drug resistance or positive family history). Finally, clinicians must be vigilant with regard to attributing neurological abnormalities to neuroleptic medication (e.g., interpreting spasticity as parkinsonian rigidity) (
85). Systemic symptoms are useful clinical pointers of an inherited leukoencephalopathy and can be used to distinguish different disorders (
Table 1).
Trigger events.
An identifiable stress-related trigger followed by an acute neuropsychiatric deterioration can point toward a diagnosis of VWMD, which warrants brain MRI. Triggers include head traumas, fever, infections, emotional distress, and other minor and major insults (
67).
Consanguinity.
The presence of consanguinity in the family of the patient should always raise a red flag for disorders inherited in an autosomal recessive fashion. These include AARS2-related leukoencephalopathy, VWMD, MLD, CTX, POLR3-related leukodystrophy, and CARASIL.
Familial psychiatric or neurological history.
Although some disorders may occur sporadically, having a positive family history for psychiatric illness may be an additional clue to suspect genetic leukoencephalopathy. Neurological diagnosis in the family is also helpful in terms of raising suspicion for a white matter disorder. For example, a family history of migraines and/or recurrent ischemic events at a young age should suggest CADASIL, CARASIL, or CARASAL. Careful consideration should be given to the diagnosis of multiple sclerosis, especially when atypical, in the family. As previously reported, adult genetic leukoencephalopathies are easily misdiagnosed as an acquired neurological disorder, and among these, multiple sclerosis is very frequent (
22). Genetic disorders that are more likely misdiagnosed as multiple sclerosis are genetic vasculopathies (CADASIL, CARASIL), ALSP, VWMD, ADLD, X-ALD (cerebral subtype), and mitochondrial disorders (
22,
50,
108–
110).
In a patient with one or more suggestive features, performing a brain MRI can provide crucial information by detecting the presence of white matter abnormalities. In general, while multifocal white matter changes evoke an acquired disorder, bilateral, confluent, and symmetric abnormalities suggest an underlying genetic etiology. Neuroradiologists and neurologists with expertise in genetic leukoencephalopathies can recognize patterns of white matter involvement that are typical of specific disorders. This is particularly true for childhood-onset genetic leukoencephalopathies. In adult forms, we are presented with two additional challenges. First, several genetic leukoencephalopathies—specifically, vascular genetic white matter disorders—present with multifocal abnormalities. Second, only a few studies have succeeded in applying an MRI pattern-recognition approach to adult genetic leukoencephalopathies because of the heterogeneity and limited specificity of patterns for each disorder (
21). The possible overlapping presence of additional acquired causes of white matter pathology (e.g., hypertension and diabetes) further complicates the task. However, some shared and recurrent MRI features can be recognized in patients with the same disorder; therefore, we adapted an MRI algorithm (
Figure 1) from those previously published to guide clinicians and radiologists when evaluating a patient with psychiatric symptoms and white matter abnormalities.
When white matter abnormalities are identified in a patient with psychiatric presentation and features suggesting a hereditary disorder, genetic assessment is mandatory. If clinically available, whole-exome sequencing should be performed. The availability of NGS-based diagnostic tests in the clinical setting has enormously accelerated the possibility of achieving a final diagnosis in patients with suspected leukoencephalopathy. Nowadays, we can sequence more than 200 genes associated with genetic leukoencephalopathies with a single test (
3,
26). Unless the clinician identifies very specific and pathognomonic signs for a definite disorder, in which case a single gene sequencing may be diagnostic, performing an NGS panel for the genes known to be associated with leukoencephalopathies has the highest yield of detecting causative mutations (
3). The advantage of whole-exome sequencing compared with NGS panels is the added possibility to identify novel genes associated with hereditary leukoencephalopathies or expand the phenotypes of known genetic disorders not previously associated with white matter abnormalities. For these reasons, it should be prioritized when clinically available. In the context of patients with undiagnosed leukoencephalopathy of probable hereditary origin, it is crucial to have access to specialized research groups in the field that can provide reanalysis and next-level investigations, such as whole-genome and RNA sequencing. On the other hand, NGS panels for dementia syndromes should include genes associated with genetic leukoencephalopathies that can present with cognitive/psychiatric involvement.
Conclusions
Early diagnosis of genetic leukoencephalopathies underlying a psychiatric presentation is of great importance, as many of these conditions are progressive and have an impact on entire families, and some may be treated with targeted therapies. In addition, patients with genetic degenerative disorders need special care that includes specific supportive therapies. It is now more crucial than ever that geneticists, neurologists, imaging experts, and psychiatrists work in multidisciplinary teams and collaborate in the investigation and management of adult patients with such disorders.
Acknowledgments
The authors thank Dr. David S. Lynch, from the University College of London Institute of Neurology, National Hospital for Neurology and Neurosurgery, London, for providing the images of AARS2-related leukodystrophy and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy.