Antibodies to neuronal surface antigens, such as the
N-methyl-
d-aspartate receptor (NMDAR), cause an autoimmune encephalitis characterized by cognitive dysfunction, psychiatric symptoms, seizures, and movement disorder (
1). Although the NMDAR antibody immunoglobulin G (IgG) isotype mediates encephalitis, NMDAR antibodies, particularly of the immunoglobulin A (IgA) and immunoglobulin M (IgM) isotypes, are also detected in patient populations in the absence of encephalitis. NMDAR antibodies of all isotypes have been shown to have pathogenic potential, with impaired glutamatergic signaling likely caused by NMDAR internalization at the synapse (
2), but the clinical significance of NMDAR antibodies outside the context of encephalitis remains unclear. However, in a number of clinical conditions, including cancer (
3), psychosis spectrum disorders (
4), stroke (
5), and postviral encephalitis (
6), IgA and IgM NMDAR antibodies have been associated with cognitive impairment. In psychosis spectrum disorders and stroke, there is also some evidence to suggest that NMDAR antibodies are associated with neuropsychiatric symptoms, such as psychosis and depression (
4,
7,
8). In addition, an association between NMDAR antibodies and psychotic symptoms in Alzheimer’s disease has been reported, but this finding has not been consistently replicated (
9,
10).
NMDAR antibodies are found in persons with dementia, particularly when the dementia subtype is atypical or unclassified, and cognitive impairment in these situations is often reversible with immunotherapy (
8,
11). However, neuronal antibodies, particularly NMDAR antibodies, are also found in well-phenotyped classical presentations of neurodegenerative disease, although it is not clear how these antibodies relate to cognitive and other neuropsychiatric symptoms (
8). This is an important question to address given the potential reversibility of antibody-mediated changes for symptoms that otherwise have few effective treatment options.
Neuropsychiatric symptoms and cognitive impairment are common among patients with PD, with more than 80% of patients developing dementia within 20 years of diagnosis (
12). The particular time course for the development of dementia is highly variable and likely reflects the contribution of many factors, such as alpha-synuclein, phosphorylated tau, and beta-amyloid (
13). The role of inflammation is also increasingly recognized in PD, including a possible role for humoral immunity to influence clinical phenotype (
14). However, studies examining the prevalence and clinical relevance of NMDAR antibodies in Lewy body disease have produced conflicting results: one study found increased numbers of serum IgA and IgM NMDAR antibodies in patients with Lewy body dementias (dementia with Lewy bodies and PD dementia) (
10), while another found no association with IgA antibodies and cognitive impairment in patients with PD, although the cognitive dysfunction exhibited by the patients in this study was modest (
15).
When NMDAR antibodies are present in the context of neurodegenerative disease, the site of autoantibody production is not clear. In autoimmune encephalitis, it is likely that ongoing peripheral germinal center reactions generate antigen-specific plasma B cells, which differentiate and access the central nervous system (CNS) to produce pathogenic IgG (
16). In neurodegenerative disease, it may be that the accelerated neuronal cell death and apoptosis lead to production of these antibodies. If NMDAR antibodies were produced following neuronal death, seropositivity might be expected to be associated with higher plasma levels of neurofilament light (NfL), a marker of neuroaxonal injury (
17), and p-tau181, a marker of phosphorylated tau (
18). Cerebral spinal fluid (CSF) levels of NfL and tau have been reported to be markedly elevated in acute NMDAR antibody encephalitis and decrease with treatment (
19,
20), but these biomarkers have not been examined in association with neuronal antibodies outside the context of encephalitis. Increased plasma NfL and p-tau181 concentrations are associated with faster cognitive decline in those with Lewy body dementias (
21–
23); thus, in measuring these biomarkers in association with NMDAR antibodies, we hope to delineate potential influences on cognitive and neuropsychiatric symptoms observed among patients with PD.
In a well-characterized prospective cohort of patients with PD, we aimed to quantify the presence of IgA, IgM, and IgG NMDAR antibodies among patients with PD compared with healthy control subjects; to explore associations between NMDAR antibody positivity and cognitive and neuropsychiatric symptoms in PD, both at baseline and longitudinally; and to investigate the relationship between NMDAR antibodies and plasma levels of p-tau181 and NfL.
Methods
Patient Cohort
Baseline plasma samples were taken from 108 patients with a clinical diagnosis of idiopathic PD who participated in the Non-motor International Longitudinal Study (NILS) at King’s College Hospital, London, and 89 healthy control subjects. The NILS is a prospective cohort study designed to assess outcomes from nonmotor symptoms in PD over time; patients are initially assessed at baseline, with plasma samples collected and clinical measurements taken. Clinical measurements of the patients are then assessed annually for up to 6 years after inclusion. All included patients had a diagnosis of PD made by clinicians who were movement disorder specialists and in accordance with internationally recognized criteria (
24). Exclusion criteria were atypical parkinsonism, age at PD onset <21 years old, insufficient clinical information, or insufficient archived plasma available for analysis. The NILS was authorized by local ethics committees. All patients provided written consent prior to study procedures, and all patient data were anonymized and coded.
Plasma samples from 89 healthy control subjects who consented were retrieved from archives at the South London and Maudsley Biomedical Research Centre, and to the extent possible, the samples were from individuals who were age- and sex-matched at the group level.
Clinical Data
Data extracted from the NILS database for patients with PD included sex, age, age at onset of PD, years of education, and Hoehn and Yahr stage (measures of PD progression) (
25), as well as scores on the Scales for Outcomes in Parkinson’s Disease (motor), with higher scores denoting better performance (
26), the Non-Motor Symptoms Scale (NMSS) for Parkinson’s Disease, with higher scores denoting greater severity or frequency of nonmotor symptoms (
27), the Hospital Anxiety and Depression Scale (HADS), with higher scores denoting a higher anxiety or depression level (
28), and the Mini-Mental State Examination (MMSE), with higher scores denoting better cognition (
29). Scales were administered by research clinicians. Psychotic symptoms were extracted from the hallucination and delusion items on the NMSS, and patients were considered to meet criteria for psychosis if they received a score
≥1 when frequency and severity were combined.
Plasma NMDAR Antibodies
All plasma samples were tested for NMDAR antibodies by using fixed cell-based assays, and testers were blinded to all clinical information. Samples were stored at −80°C until assayed. IgA, IgM, and IgG NMDAR antibodies were detected with a recombinant cell line transfected with an expression construct for the reactive receptor subunit NR1 utilizing indirect immunofluorescence (Euroimmun NMDAR kit, Lübeck, Germany) as described previously (
30). Transfected human embryonic kidney 293 cells were incubated with a plasma dilution of 1:10 from each patient; fluorescein isothiocyanate-conjugated secondary antibodies (IgG, IgA, and IgM) were then added, and antibody binding was measured with immunofluorescence.
Plasma NfL Concentration
Plasma NfL concentrations were measured with a commercially available NF-Light kit on a single-molecule array Simoa HD-1 analyzer (Quanterix Corp., Billerica, Mass.) for 143 samples (PD group: N=105; healthy control group: N=38) at the UK Dementia Research Institute Fluid Biomarkers Laboratory in a single round of experiments with one batch of reagents. Testers were blinded to the samples, and 91% (N=130) of samples were measured in duplicate (there was insufficient material available for 13 samples; patients with PD: N=6, healthy control subjects: N=7). Intra-assay coefficients of variation were <5%. The limit of detection was 0.038 pg/mL, and the lower limit of quantification was 0.174 pg/mL.
Plasma p-tau181 Concentration
Plasma phosphorylated tau was measured in 104 PD samples at King’s College London with the commercially available Simoa pTau-181 Advantage V2 kit (Quanterix Corp.). Plasma was diluted to 1:4 and read on an HD-1 analyzer. Data acquisition spanned five analytical runs; the lower limit of quantification for this assay was 0.127 pg/mL, and the coefficient variation for interassay and intra-assay variability was 7.51% and 7.69%, respectively.
Statistical Analysis
Healthy control subjects and patients with PD were compared across age, gender, antibody status, and NfL concentration. NMDAR antibody seropositive and seronegative PD patients were compared with regard to neuropsychiatric symptoms, cognitive outcomes, and functional disability, both at baseline and during the follow-up period. Continuous variables were compared by using independent t tests or Mann-Whitney U tests, dependent on the distribution of the data. Categorical data were analyzed with chi-square test or Fisher’s exact test.
To assess the longitudinal effects of NMDAR antibodies, the annual decline in MMSE scores over the follow-up period was calculated. Linear regression was performed for annual decline in scores across subgroups with positive or negative NMDAR antibody status, and age, sex, years of education, follow-up duration, disease duration (months), and p-tau181 and NfL concentrations were used as covariates. Cognitive impairment was also classified dichotomously; patients with an MMSE score <26 were classified as cognitively impaired.
To achieve normality, NfL and p-tau181 concentrations were log-transformed. Within the patients with PD, linear regression was used to analyze log NfL and log p-tau181 concentrations across subgroups with positive or negative NMDAR antibody status, with age used as a covariate.
The significance threshold was set at a p value <0.05, and where applicable, values are reported for two-tailed tests. Significance values were uncorrected for multiple comparisons due to the exploratory nature of the analyses. Statistical analyses were conducted with Stata, version 16.0.
Results
Participants
A total of 108 patients with PD (25% female; mean±SD age=63.7±11.9 years) and 89 healthy control subjects (34% female; mean age=54.2±17.0 years) were included (
Table 1); healthy control subjects were significantly younger than patients with PD (p<0.001). As reported in
Table 2, the baseline mean Hoehn and Yahr Scale score for patients with PD who were negative for NMDAR antibodies was 2.34±0.76. The duration of PD at baseline was highly variable, with a median of 6.8 years (range 1–32).
Sixty-one (56%) patients with PD (28% female, mean age=63.3±11.6 years) were followed longitudinally (mean duration=3.69±1.73 years; median=3.99 years, range 0.71–6.61 years). The baseline mean MMSE score for these patients was 28.8±2.4, which declined by a mean of 0.60±1.24 points annually during the follow-up period.
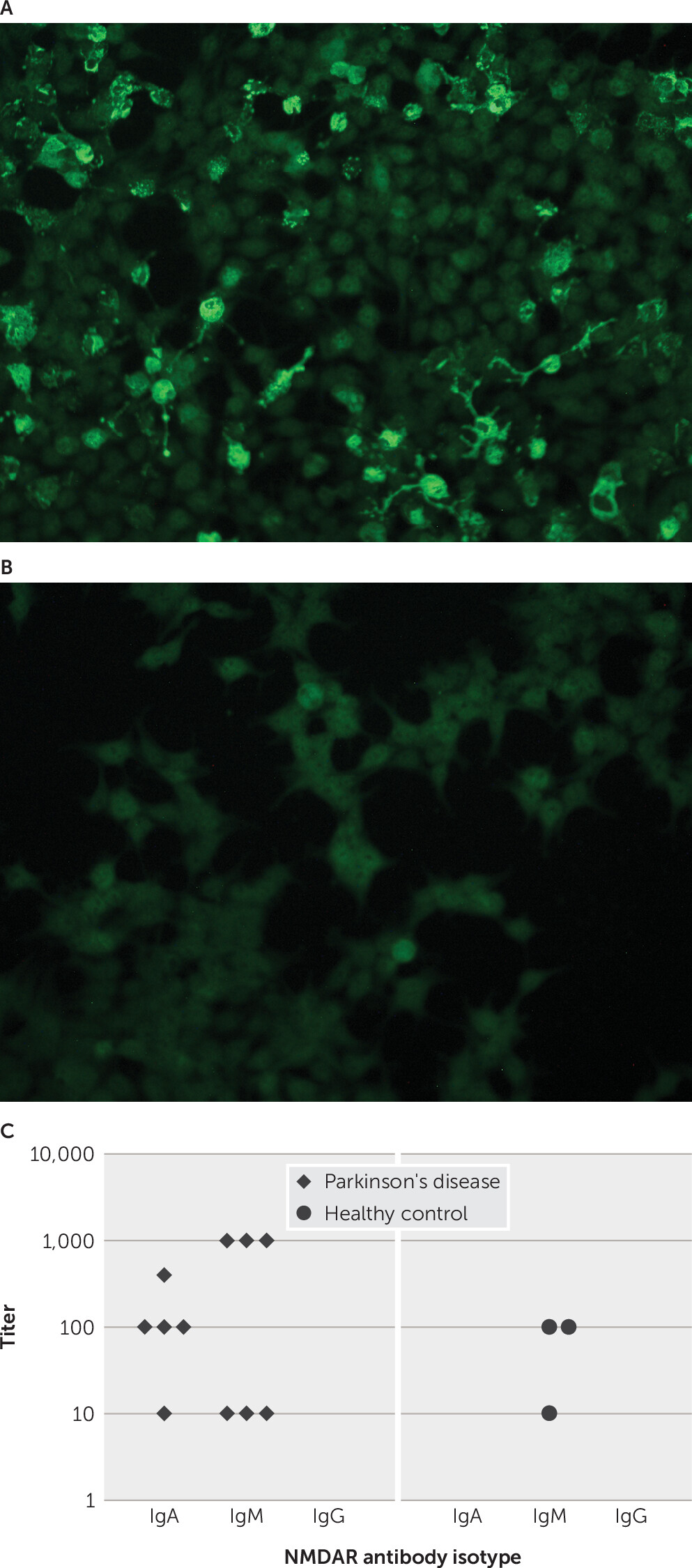
Plasma NMDAR Antibody Positivity Among PD Patients and Healthy Control Subjects
Ten (9%) patients in the PD group tested positive for NMDAR antibodies (IgA, N=5; IgM, N=6; IgG, N=0); one of these patients had both IgA and IgM NMDAR antibodies. Three healthy control subjects (3.3%) tested positive for NMDAR antibodies, all with an IgM isotype antibody. IgA NMDAR antibodies were significantly more common among PD patients than healthy control subjects (χ
2=4.23, df=1, p=0.04) (
Figure 1); IgA NMDAR antibodies were not detected in any healthy control subject (
Figure 1C)
. There was no association between NMDAR antibody positivity and age (t=−0.41, df=195, p=0.68).
No Demographic Differences Among the Patients With PD and NMDAR Antibody Positivity
No differences in age (t=0.39, df=106, p=0.69), gender (χ
2=0.15, df=1, p=0.70), age at PD onset (t=0.51, df=106, p=0.61), or follow-up duration (t=−0.78, df=59, p=0.44) were observed between the patients with PD and NMDAR antibody positivity or negativity. There was also no difference in motor symptoms (t=0.13, df=81, p=0.89) or degree of disease progression (Hoehn and Yahr Scale) (Mann-Whitney U=0.12, p=0.93) based on antibody status (
Table 2).
Cognitive Associations of NMDAR Antibody Positivity Among the Patients With PD
At baseline, there was no difference in MMSE scores based on antibody status (Mann-Whitney U=0.29, p=0.77). However, antibody status was a significant predictor of annual decline in MMSE scores in the linear regression analysis that was adjusted for age, sex, years of education, p-tau181, NfL, disease duration, and follow-up duration (adjusted R
2=0.26, p=0.01). The mean annual decline in scores was 1.36±2.55 for the antibody-positive subgroup and 0.50±0.96 for the antibody-negative subgroup. During the course of the follow-up period, 13 patients with PD (21%) showed evidence of cognitive impairment (MMSE score <26); of these, 31% (N=4) were seropositive for NMDAR antibodies. For those followed longitudinally, 57% (N=4) of antibody-positive patients showed evidence of cognitive impairment at some time point in the course of the study compared with 17% (N=9) of antibody-negative patients (χ
2=6.05, df=1, p=0.014) (
Table 3).
Neuropsychiatric Outcomes With NMDAR Antibody Positivity Among the Patients With PD
During the course of the study, 37 (35%) patients with PD reported psychotic symptoms (psychosis at baseline, N=22; emergent psychosis during the follow-up period, N=15). There was no relationship between psychotic symptoms and NMDAR antibody status (χ2=0.31, df=1, p=0.58).
Baseline HADS scores showed high morbidity of depression and anxiety among the patients with PD (mean=10.4±7.41). Baseline HADS scores were not significantly different across the antibody-positive and antibody-negative subgroups (Mann-Whitney U=0.55, p=0.59). The mean change in the HADS score during the follow-up period was an increase of 6.56±6.34 points, and this was not significantly different in antibody-positive and antibody-negative patients with PD (Mann-Whitney U=−0.93, p=0.35).
Plasma NfL and p-tau181 Concentrations With NMDAR Antibody Positivity
The mean NfL concentration across all samples was 25.3±17.4 pg/mL. NfL concentrations were lower in samples from healthy control subjects than in samples from PD patients, but the difference was not significant (log NfL t=−0.74, df=141, p=0.46; see
Table 1). Among the patients with PD, there was no association between NfL concentrations and antibody status in an age-adjusted linear regression analysis (R
2=0.14, p=0.98). In the patients with PD, the mean plasma p-tau181 concentration was 2.32±1.36 pg/ml. There was no association between plasma p-tau181 levels and antibody status in the linear regression analysis that was adjusted for age (adjusted R
2=0.10, p=0.32).
Discussion
In one of the first studies to investigate the possible longitudinal implications of NMDAR antibodies in neurodegenerative disease, we found increased frequencies of IgA NMDAR antibodies in patients with PD, and seropositive patients showed significantly greater cognitive impairment over time. More than one-half (57%) of all seropositive patients with PD were cognitively impaired during the study, while only 17% of seronegative patients showed evidence of cognitive impairment. Seropositive patients showed significantly greater annual cognitive decline irrespective of p-tau181 and NfL concentrations and other clinical and demographic variables. These findings suggest that any antibody-associated cognitive impairment occurs independent of these coexisting neurodegenerative processes.
NMDAR Antibody Isotype
When NMDAR antibodies are detected outside the context of encephalitis, they are primarily of the IgA and IgM isotypes. Consistent with previous studies, we detected only IgA and IgM antibodies, with IgA NMDAR antibodies found in patients with PD and not in healthy control subjects. There is some disagreement regarding the clinical relevance of IgA and IgM antibodies; while in vitro all NMDAR antibody isotypes have the potential to cause receptor internalization and dysfunction of glutamatergic signaling (
2), a second study found a minority reacted with live neurons with no observed decrease in synaptic or extrasynaptic NMDARs (
31). However, IgA antibodies have been implicated clinically with slowly progressive cognitive impairment that is reversible with immunotherapy (
32). IgA NMDAR seropositivity has also been associated with cognitive dysfunction in a number of cancers, with impairments proportionate to antibody titer and the degree of blood-brain barrier disruption (
3,
33). Similarly, in a case-control study of people at clinical high risk of psychosis, IgA NMDAR seropositivity was associated with significantly lower IQ and impaired performance on the Rey Auditory Verbal Learning Test (
4). These impairments mirror the findings in our study, and therefore it seems possible that these antibody-associated effects are not specific to a particular disorder, with cognitive impairment observed across a range of clinical populations. However, it is also possible that the antibody-associated cognitive impairment represents a nonspecific factor, for example, indicative of patients who have a greater degree of inflammation, which could drive clinical outcomes.
NfL and p-tau181 Concentrations
Plasma NfL is a biomarker of neuroaxonal injury that is not specific for any one disease (
17); plasma p-tau is an accurate marker of phosphorylated tau (
18). We found that NfL concentrations did not significantly differ between patients with PD and healthy control subjects, which is consistent with previous studies showing no increase in NfL concentration in PD (
34). Furthermore, we found no association between NfL or p-tau181 levels and NMDAR antibodies in the patients with PD, which suggests that these antibodies are present independent of the coexisting neurodegeneration and neuroaxonal injury in PD, consistent with the presence of NMDAR antibodies not being related to the degree of neurodegeneration in PD.
NMDAR antibodies are known to target the NR1 subunit of the receptor, causing internalization and reduction of NMDAR-mediated currents without causing cell death (
35). However, in acute IgG-mediated NMDAR encephalitis, there is a significant increase in CSF NfL concentration, which suggests that there are pathological processes extending beyond the reversible internalization of NMDAR (
19). If IgA or IgM NMDAR antibodies are exerting an effect in PD, these effects may more likely be restricted to the synapse and distinct from the inflammatory processes known to occur in IgG NMDAR encephalitis, and thus without axonal injury and subsequent increases in NfL levels (
36). Indeed, chronic presence of serum NMDAR antibodies causes cognitive dysfunction, indicated by impairments in spatial working memory and novelty detection in otherwise healthy mice (
37). Indeed, most passive immunization animal models of NMDAR encephalitis show impairments restricted to cognition, without the diverse symptomology and CNS inflammation associated with NMDAR encephalitis in humans (
38). This is not unlike the modest cognitive impairments observed in the present study; it seems likely that if NMDAR antibodies do have a causative role in neurodegenerative disorders, then this role is restricted to the synapse and does not influence the additional inflammatory processes inherent to IgG-mediated NMDAR encephalitis (
36,
38,
39).
Clinical Relevance in PD
While the etiology of PD remains to be elucidated, there is accumulating evidence that neuroinflammation plays a role early in the disease process (
40). Up to 80% of people with PD develop dementia, but there is significant heterogeneity in the clinical course and time to develop symptoms. This heterogeneity likely reflects the interplay of many divergent biological factors, of which neuronal autoantibodies could be one. Several prognostic biomarkers have recently been identified as being associated with cognitive decline in PD, with MRI measures of cortical atrophy and CSF and blood biomarkers of Alzheimer’s disease pathology holding particular promise (
41). Indeed, plasma p-tau181 is an accurate marker of phosphorylated tau associated with cognitive decline in neurodegenerative dementia (
42,
43). By showing NMDAR antibodies to be associated with cognitive decline, independent of p-tau181, we hope to address concerns that the cognitive decline seen in patients with seropositive PD exclusively relates to other neurodegenerative processes in PD. Future studies could include a wider array of biomarkers to assess other pathologies implicated in cognitive impairment.
Limitations
There were several limitations to this cohort study. Some of the group sizes were small, primarily due to the 44% loss during the follow-up period and the small number of samples with detectable NMDAR antibodies. Furthermore, although efforts were made to match patients in the PD group and healthy control group according to age and gender, the limited availability of samples from healthy control subjects led to a significantly younger healthy control group than PD group. No association was found between NMDAR antibody positivity and age, and thus we do not believe that this age difference affects the overall validity of the results. We also adjusted for age in all regression models.
Not all patients were followed up for the same duration after inclusion in the study; therefore, to minimize missingness, annual decline in MMSE scores was calculated from the MMSE scores available throughout the follow-up period. There were no significant differences in follow-up durations between the seropositive and seronegative groups, making this an unlikely confounder, and follow-up duration was also included as a covariate in the regression model. The calculated annual decline in MMSE scores demonstrated a difference of almost 1 point between the antibody-positive and antibody-negative subgroups annually, but this difference did not reach statistical significance outside the regression model due to the small group sizes in this analysis.
We used the MMSE to assess cognition, which is known to be less sensitive than the Montreal Cognitive Assessment (MoCA) in PD. However, the MMSE and MoCA are equally sensitive in measuring cognitive change over time in PD, and therefore this should not affect the validity of our results (
44). Psychosis, depression, and anxiety were assessed with the NMSS and HADS. While these scales provide a broad assessment of these symptoms, they do not comprehensively cover neuropsychiatric symptoms and are less detailed than other scales. In future studies, investigators could consider including the Neuropsychiatric Inventory or Movement Disorder Society Nonmotor Rating Scale in their assessments.
CSF samples were not routinely collected in this prospective longitudinal cohort study, and thus our analyses were restricted to plasma samples. However, while CSF analysis remains the gold standard for detection in autoimmune encephalitis, this does not negate the potential clinical relevance of NMDAR antibodies found in plasma. Furthermore, previous studies have shown that neuronal antibodies are not commonly found in the CSF of persons with dementia (
10).
Conclusions
In this longitudinal cohort study, we found NMDAR antibody seropositivity to be associated with greater annual cognitive decline in patients with PD. Further investigation is needed to explore whether NMDAR antibodies have a causative role in the cognitive decline in PD, but the reversibility of immune-mediated mechanisms offers exciting potential for the development of therapeutic interventions.
Acknowledgments
The authors thank the NIHR BioResource volunteers for their participation, as well as the NIHR BioResource centers, NHS trusts, and their staff for their contributions. The authors also thank NIHR, NHS Blood and Transplant, and Health Data Research UK.