Surprisingly, very few studies have looked at CRF as a possible blood biomarker for PTSD in humans. A study in cerebrospinal fluid (CSF) showed that patients with PTSD with secondary psychotic symptoms had higher CRF compared to PTSD without psychosis, and healthy subjects (
14), implicating abnormalities in the secretion of CRF with the production of secondary psychotic symptoms in PTSD. A few studies in PTSD patients reported high levels of CRF in CSF (
15), while diurnal plasma cortisol levels on average were decreased (
16). One study on plasma CRF showed an increase in PTSD (
17). However, the cohorts were of only 30 subjects, and 13 out of 31 PTSD patients were diagnosed with a current depressive episode (
16). To our knowledge, no previous studies have proposed serum CRF to have diagnostic potential for the differentiation of PTSD and TBI, and studies on the differentiation of PTSD and TBI have been mostly limited to functional neuroimaging (
18,
19).
Diagnosis of PTSD and TBI is most often determined from clinical interviews or self‐report assessments, both of which are prone to distortions and depend on the willingness of patients to discuss their traumatic experiences and related symptoms. Easily accessible blood biomarkers would aid in the understanding, diagnosis and treatment of both PTSD and TBI.
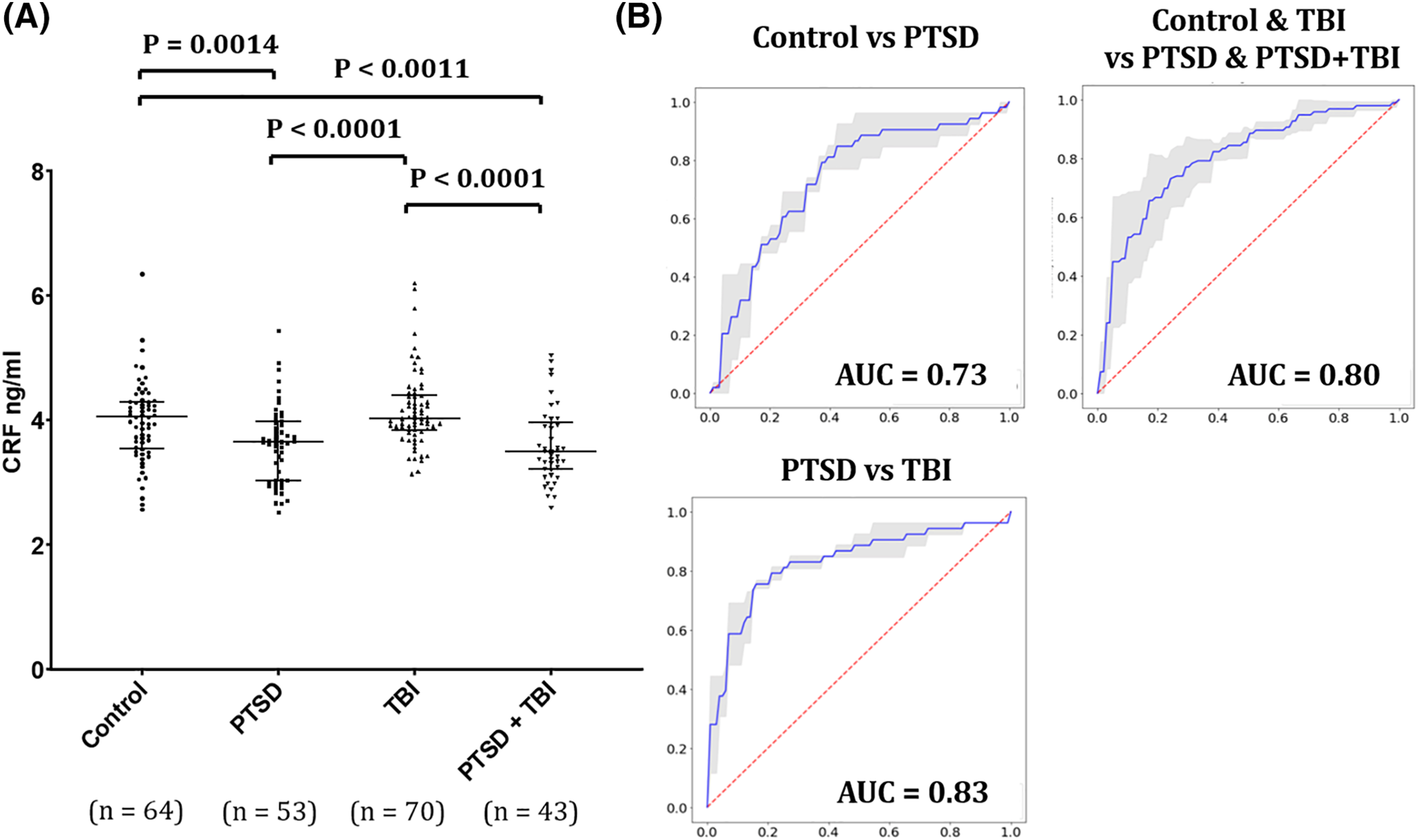
Here, we report that serum CRF is significantly lower in a large group of combat veterans with PTSD compared to healthy veterans as well as veterans with evidence of past TBI defined by loss of consciousness (LOC).
Discussion
The HPA axis is one of the major neuro‐hormonal systems mediating the stress response in the human body and neuroendocrine studies consistently provide evidence for altered HPA activity in PTSD. (
23) The present report points for the first time to reduced serum CRF as a promising biomarker for differentiating PTSD from health individuals and from subjects with chronic TBI. We found that CRF levels were lower in veterans with PTSD and PTSD + TBI, compared to healthy veterans and veterans with a history of TBI, with moderate to large effect sizes. Our findings suggest that, if validated, serum CRF may be considered for membership in future blood biomarker panels for PTSD.
The activity of the HPA axis is regulated by multiple sympathetic and parasympathetic inputs and limbic circuits (e.g., amygdala, hippocampus, and medial prefrontal cortex), directly or indirectly innervating the PVN of the hypothalamus (
24). In healthy subjects, integrated brain circuits trigger the parvocellular neurons of the PVN to release infundibular CRF, which stimulates the release of ACTH from corticotropic cells of the pituitary. ACTH reaches the bloodstream and triggers the systemic release of glucocorticoids by the adrenals. The classic view of the HPA axis involves activation of cortisol response under acute stress conditions. However, recent studies show a complex modulation including enhanced feedback inhibitory systems in chronic stress and addiction (
9,
10).
Animal studies demonstrate that HPA axis responsivity to acute stressors is altered in animals that have previously been stressed, suggesting a long‐term negative feedback modulation of the HPA axis in response to chronic stress (
25). While exposure to severe stressors causes long term dysregulation of resting state and stress‐induced activation of the HPA axis (
26,
27,
28), the mechanisms of such modulation are complex and unclear, and both increased and blunted cortisol response are observed depending on the intensity and chronicity of the stressors. (
29) Early biomarkers studies in humans typically focused on plasma or saliva cortisol determinations and reported, as a general rule, increased levels in response to acute stress, (
11,
12) suggesting that PTSD biology was compatible with a stress response, associated with increased CRF levels, catecholamine depletion within the central nervous system, and reduced hippocampal volume. (
30) However, the activation of the HPA axis may vary with duration of PTSD. Cross‐sectional and prospective studies are suggestive of a hypoactive HPA axis in PTSD (
31). Indeed, lower urinary and plasma cortisol levels were detected in chronic PTSD patients compared to non‐PTSD trauma survivors and controls. (
30) This was confirmed by experiments using dexamethasone as an HPA axis inhibitor in different cohorts. A study by Yehuda and colleagues showed lower post‐DEX ACTH levels in deployed veterans than in a non‐deployed veteran group, suggesting a link between post‐deployment symptoms and chronic stress (
32). In our study, we observed a clear reduction in CRF levels in the chronic PTSD group that was also detected in the PTSD + TBI group, but not in the TBI alone, suggesting that the effect may mediated by a biological mechanism related to PTSD and independent from TBI. Our PTSD cohort included both subjects with (n = 42) and without (n = 54) current depression. We didn't observe significant differences in these two subgroups, suggesting that the lower levels of CRF are driven by PTSD and not by the comorbid depression. Additionally, the correlation of serum CRF with BDI was not significant, except for the PTSD + TBI group, in which a positive correlation was observed between BDI and CRF (r = 0.555; P = 0.001). This further confirms that our findings of decreased CRF in PTSD are unrelated to depressive symptoms, as in this group higher CRF levels associated with a higher depression score. To the best of our knowledge, this serum CRF reduction in PTSD compared to controls and TBI subjects has not been described before and is in line with a more recent perspective of PTSD biology that involves a chronic stress state and enhanced negative feedback of the HPA axis. This effect can also be related to the higher sensitivity of glucocorticoid receptors in peripheral tissues that has been previously observed in animal models (
30,
33,
34). Our results are also in line with a recent study, which found a reduction in pituitary gland size and altered function in PTSD. That study reported discordant pituitary corticotropin function and pituitary volume in PTSD subjects compared with intact HPA feedback and association of pituitary volume with ACTH levels in healthy control subjects (
35).
The observed low serum CRF in chronic PTSD patients may be also consistent with elevated brain CRF utilization, increased CRF metabolism, increased degradation of CRF, or reduction in the CNS secretion of CRF (
36).
Alternatively, low serum CRF may represent a risk factor for PTSD. This possibility should be further explored in longitudinal studies with available blood obtained from participants pre‐ and post‐deployment or pre‐ and post‐trauma exposure in civilians.
Since psychotropic medications such as benzodiazepines and antidepressants may influence CRF levels (
37), we have compared PTSD subjects (including PTSD + TBI) under benzodiazepines and antidepressant treatments with those without drug treatment. We found that the levels of serum CRF did not differ in the two groups (p = 0.472), confirming that the CRF reduction in PTSD is not due to the use of these medications.
Serum CRF is known to be increased in multiple conditions, including fibromyalgia, mastocytosis, psoriasis, obesity, and pregnancy (released by the placenta) (
38,
39,
40,
41). All participants of our study, including the controls, were subjected to a thorough clinical interview and all possible current heath conditions were reported. None of the participants were found to have current fibromyalgia, mastocytosis or psoriasis at the time of the interview and/or blood draw. In addition, we excluded from this study any pregnant subjects.
Low levels of CRF are also observed in tertiary (hypothalamic) adrenal disease (CRF deficiency) (
42). Both low and elevated CRF levels have been shown in people with Alzheimer's disease (
43,
44), and some scientists suspect that a lack of CRF might cause chronic fatigue syndrome (
45). However, further research is needed in both these topics to confirm these findings.
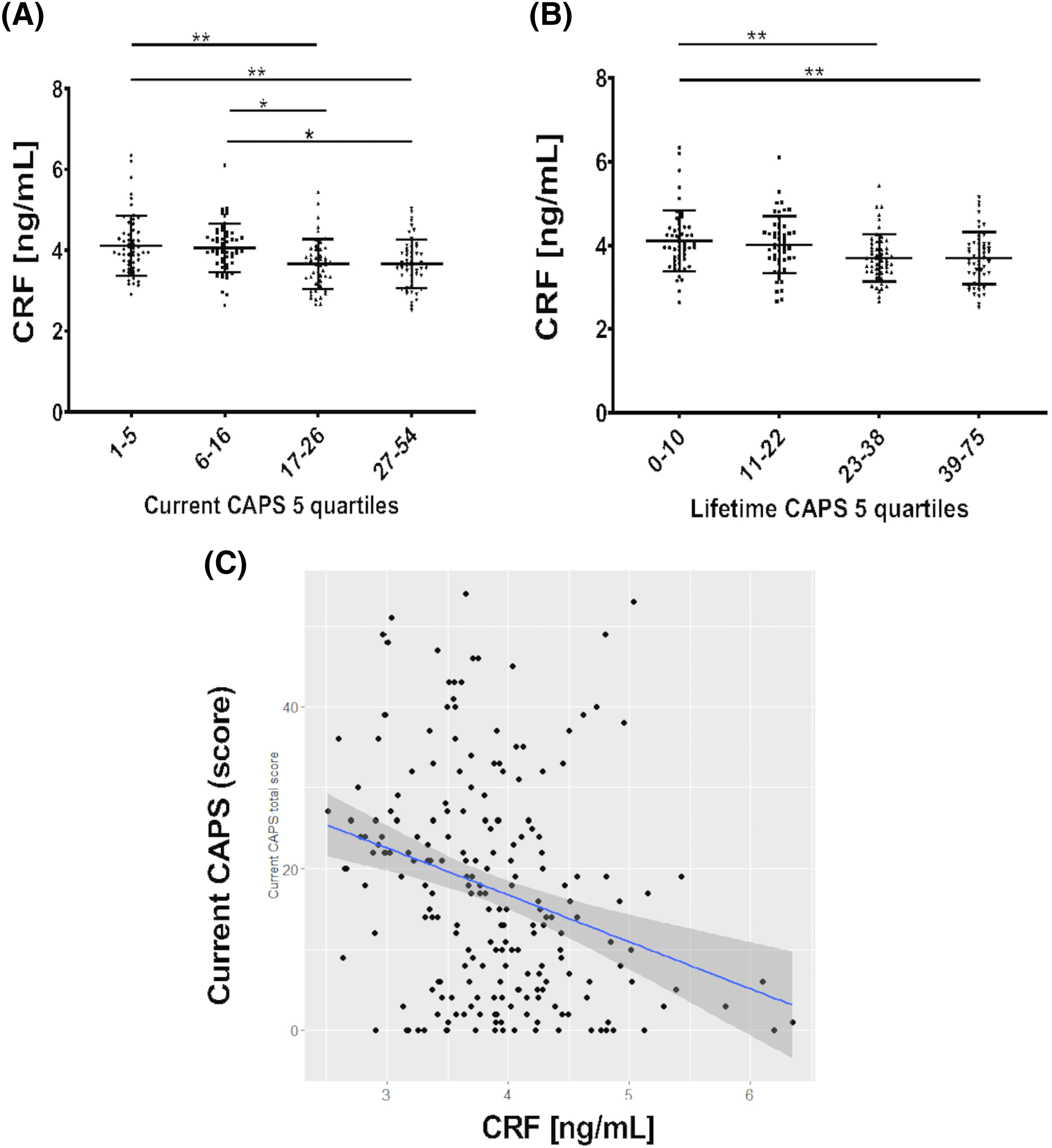
We detected lower CRF levels in subjects within the higher CAPS scores quartiles, for both current and lifetime CAPS (
Figure 2), and a moderate negative correlation between CRF and CAPS‐5 in the full group. However, there was no correlation between CAPS‐5 and CRF within the PTSD group, suggesting that the relationship with CRF is likely driven by the differences in CAPS scores between controls and PTSD subjects and is not directly related to the severity of symptoms within the PTSD patients. Further, there was no correlation between CRF and the individual CAPS‐5 symptom clusters (B, C, D or E) in the PTSD group, or between CRF and PSQI.
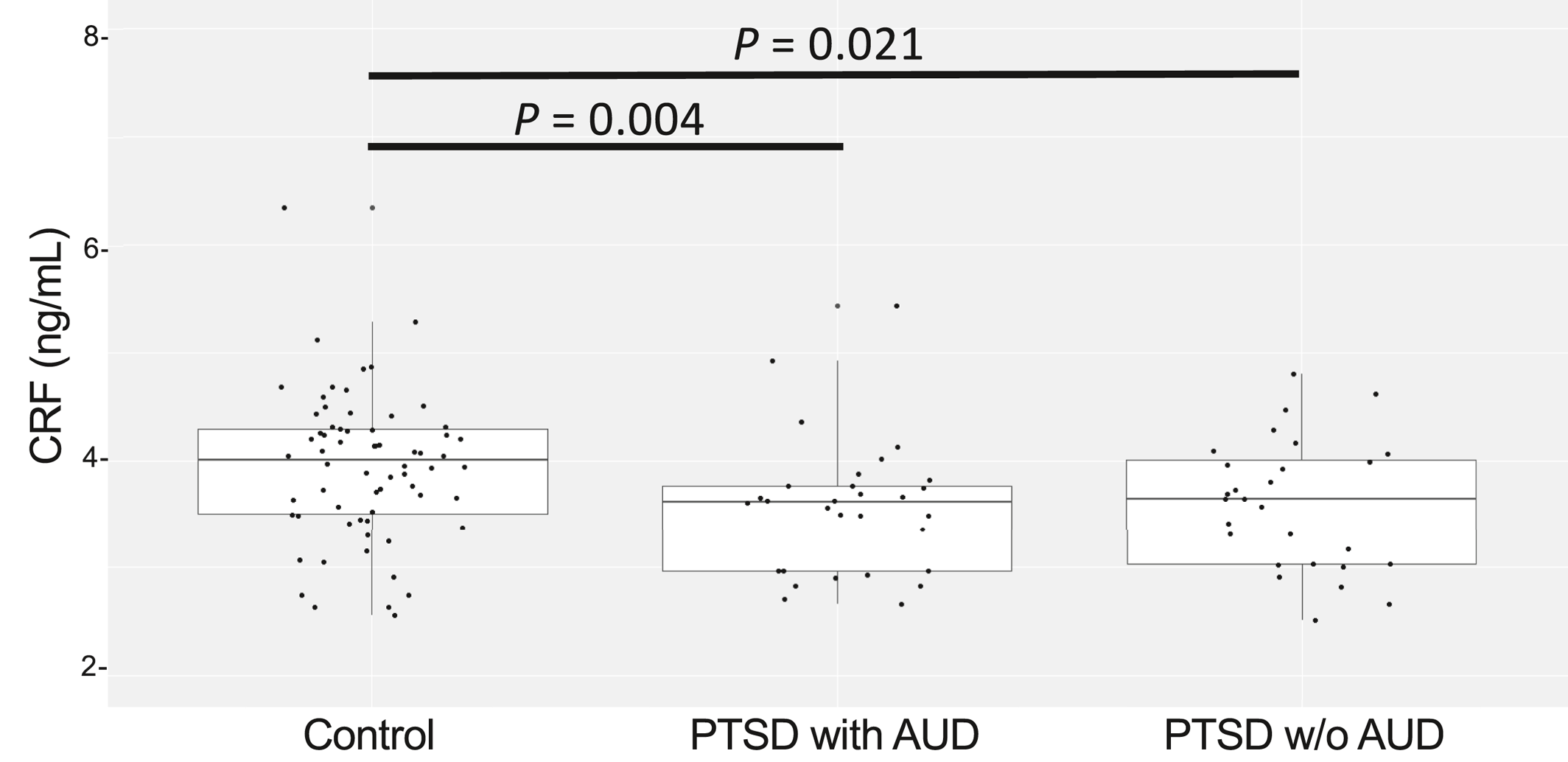
The reduction in CRF levels had a larger effect size in PTSD patients with reported moderate or severe alcohol use when compared to controls, likely due to the lower variability of CRF levels in the PTSD + AUD group. Alcohol abuse can reach a prevalence of more than 50% in PTSD patients (
46,
47,
48) and it is known to be associated with more severe PTSD symptoms and higher levels of cognitive disturbances. Although AUD was not the main focus of this study, which did not include participants with AUD in absence of PTSD, the analysis of HPA axis mediators such as CRF in comorbid PTSD and AUD as well as in AUD alone warrants further attention for a potential biological interaction.
Serum cortisol levels measured in a subset of our subjects (N = 83) did not show significant group differences (Control vs. PTSD: p = 0.46; Control vs. PTSD + TBI: p = 0.33; PTSD vs. PTSD + TBI: p = 0.95), in line with a meta‐analysis in which, across 37 studies, 828 people with PTSD and 800 controls did not differ in cortisol levels. (
49) The lack of difference persisted in this meta‐analysis when considering only studies analyzing plasma or serum cortisol. (
49).
Although the brain is the main source of CRF production related to stress, CRF is released from the PVN neurons that terminate in the median eminence, a region with fenestrated blood vessels lacking the tight junctions characteristic of the BBB. Here, CRF is released into the peripheral circulation as a hormone. Peripheral CRF (or CRH) plays multiple roles associated to the stress response. For example, CRF released from the brain into the periphery during stress responses induces a colonic response by activating colonic CRF receptors (
50). These and other peripheral mechanisms are mediated by central stress, as can be displayed by the “gut wrenching” and nauseous feelings and abdominal pain during or following emotional stress.
Hence, although due to the nature of our samples we cannot demonstrate that serum CRF levels correlate with those in the brain in our cohort, there is evidence indicating that peripheral CRF levels can be indicative of CNS perturbations, and associated to emotional disturbances and HPA axis responses. Previous research also suggests that a major component of plasma CRF is of hypothalamic origin, however, other extrahypothalamic tissues cannot be ruled out as a minor source of plasma CRF (
51). Other studies have shown lower levels of peripheral CRF (plasma) and an increase in PTSD (
17). However, the study did not control for depression, and patients with severe depression were almost half of the PTSD group in this study. We have observed an opposite direction of the correlation in depression, with an increased CRF correlating with increased depression scores (BDI) in the PTSD + TBI group. In addition, this study used an RIA assay, while we use EIA. Differences in apparent concentrations are conceivable due to the inherent technical differences of these assays. The higher overall concentration found with the commercial EIA assay we used, compared to other previously used RIA and Mass spectrometry assays for CRF is a limitation of our study, and the results should be confirmed in the future employing mass spectrometry, RIA, or other quantitative types of analysis to confirm concentrations. High backgrounds of the EIA assay, as well as possible detection of both bound and unbound CRF are possible reasons. Nevertheless, the strong effect size of our group differences in serum CRF (in controls vs PTSD and PTSD + TBI groups) reinforces the confidence that these results are biologically important, independently from possible assay‐dependent variability. We believe that the present data need to be reported, so that additional studies can focus on serum CRF determinations in PTSD and PTSD + TBI populations using multiple different assays.
The present report has some limitations that will need to be explored in future studies. First, due to the veteran nature of our study sample, to validate CRF as a biomarker for PTSD results will need to be confirmed in separate cohorts of civilian subjects. Second, our study included relatively young veterans with a homogeneous age (average 32.54) and future studies should explore if results hold in elderly subjects and if CRF levels are related to age. Third, it is important to consider that sex can affect HPA axis activity, as established by multiple animal and human studies which highlight an effect of sex hormones on stress responses (
52,
53). Our sample of Iraq and Afghanistan veterans consisted of 7.88% female subjects. Therefore, results will need to be confirmed in validation cohorts comprising a balanced number of men and women and in studies of female veterans and civilians with PTSD and TBI. Additional limitations are the lack of cortisol level measurments in all patients and the lack of a group of AUD participants without PTSD. The relationships between alcohol use and serum CRF levels as well as circadian rhythm and CRF levels should be explored in future studies. Additionally, our study did not account for potential binding of CRF to CRF‐binding protein in plasma which may impact CRF soluble levels. Future studies will be planned to assess these molecular events. Finally, longitudinal studies analyzing CRF levels before, immediately after trauma, and in chronic PTSD within the same patients, would help understanding if the lower CRF levels found in PTSD subjects are the result of a chronic stress state, or if lower CRF is an inherent characteristic of certain individuals, which constitutes a risk factor for PTSD. Studies will need to further confirm if the observed results are due, at least in part, to changes in CRF of peripheral origin, since CRF is known to be produced by other peripheral sources including immune and endothelial cells, the adrenal medulla, testes, ovaries, GI tract, pancreas, and the myometrium.