Depression is among the top five leading causes of disability and disease burden throughout the world (
1). Across the life span, stressful life events that involve threat, loss, humiliation, or defeat influence the onset and course of depression (
2–
5). However, not all people who encounter a stressful life experience succumb to its depressogenic effect. Diathesis-stress theories of depression predict that individuals’ sensitivity to stressful events depends on their genetic makeup (
6,
7). Behavioral genetics research supports this prediction, documenting that the risk of depression after a stressful event is elevated among people who are at high genetic risk and diminished among those at low genetic risk (
8). However, whether specific genes exacerbate or buffer the effect of stressful life events on depression is unknown. In this study, a functional polymorphism in the promoter region of the serotonin transporter gene (
SLC6A4) was used to characterize genetic vulnerability to depression and to test whether 5-HTT gene variation moderates the influence of life stress on depression.
The serotonin system provides a logical source of candidate genes for depression, because this system is the target of selective serotonin reuptake–inhibitor drugs that are effective in treating depression (
9). The serotonin transporter has received particular attention because it is involved in the reuptake of serotonin at brain synapses (
10). The promoter activity of the 5-HTT gene, located on 17q11.2, is modified by sequence elements within the proximal 5’ regulatory region, designated the 5-HTT gene-linked polymorphic region (5-HTTLPR). The short (“s”) allele in the 5-HTTLPR is associated with lower transcriptional efficiency of the promoter compared with the long (“l”) allele (
11).
Evidence for an association between the short promoter variant and depression is inconclusive (
12). Although the 5-HTT gene may not be directly associated with depression, it could moderate the serotonergic response to stress. Three lines of experimental research suggest this hypothesis of a gene-by-environment (G × E) interaction. First, in mice with disrupted 5-HTT, homozygous and heterozygous (5-HTT −/− and +/−) strains exhibited more fearful behavior and greater increases in the stress hormone (plasma) adrenocorticotropin in response to stress compared to homozygous (5-HTT +/+) controls, but in the absence of stress no differences related to genotype were observed (
13). Second, in rhesus macaques, whose length variation of the 5-HTTLPR is analogous to that of humans, the short allele is associated with decreased seroto-nergic function [lower cerebrospinal fluid (CSF) 5-hydroxyindoleacetic acid concentrations] among monkeys reared in stressful conditions but not among normally reared monkeys (
14). Third, human neuroimaging research suggests that the stress response is mediated by variations in the 5-HTTLPR. Humans with one or two copies of the s allele exhibit greater amygdala neuronal activity to fearful stimuli compared to individuals homozygous for the l allele (
15). Taken together, these findings suggest the hypothesis that variations in the 5-HTT gene moderate psychopathological reactions to stressful experiences.
We tested this G × E hypothesis among members of the Dunedin Multidisciplinary Health and Development Study (
16). This representative birth cohort of 1037 children (52% male) has been assessed at ages 3, 5, 7, 9, 11, 13, 15, 18, and 21 and was virtually intact (96%) at the age of 26 years. A total of 847 Caucasian non-Maori study members, without stratification confounds, were divided into three groups on the basis of their 5-HTTLPR genotype (
11): those with two copies of the s allele (s/s homozygotes;
n=147; 17%), those with one copy of the s allele (s/l heterozygotes;
n=435; 51%), and those with two copies of the l allele (l/l homozygotes;
n=265; 31%). There was no difference in genotype frequencies between the sexes [χ
2(2<)=0.02,
P=0.99]. Stressful life events occurring after the 21st birthday and before the 26th birthday were assessed with the aid of a life-history calendar (
17), a highly reliable method for ascertaining life-event histories (
18). The 14 events included employment, financial, housing, health, and relationship stressors. Thirty percent of the study members experienced no stressful life events; 25% experienced one event; 20%, two events; 11%, three events; and 15%, four or more events. There were no significant differences between the three genotype groups in the number of life events they experienced,
F(2,846)=0.56,
P=0.59, suggesting that 5-HTTLPR genotype did not influence exposure to stressful life events.
Study members were assessed for past-year depression at age 26 with the use of the Diagnostic Interview Schedule (
19), which yields a quantitative measure of depressive symptoms and a categorical diagnosis of a major depressive episode according to
Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) criteria (
20). 17% of study members (58% female versus 42% male; odds ratio=1.6; 95% confidence interval from 1.1 to 2.2) met criteria for a past-year major depressive episode, which is comparable to age and sex prevalence rates observed in U.S. epidemiological studies (
21). In addition, 3% of the study members reported past-year suicide attempts or recurrent thoughts about suicide in the context of a depressive episode. We also collected informant reports about symptoms of depression for 96% of study members at age 26 by mailing a brief questionnaire to persons nominated by each study member as “someone who knows you well.”
We used a moderated regression framework (
22), with sex as a covariate, to test the association between depression and (i) 5-HTTLPR genotype, (ii) stressful life events, and (iii) their interaction (table S1). The interaction between 5-HTTLPR and life events showed that the effect of life events on self-reports of depression symptoms at age 26 was significantly stronger (
P=0.02) among individuals carrying an s allele than among l/l homozygotes (Fig. 1A). We further tested whether life events could predict within-individual increases in depression symptoms over time among individuals with an s allele by statistically controlling for the baseline number of depressive symptoms they had before the life events occurred (table S1). The significant interaction (
P=0.05) showed that individuals carrying an s allele whose life events occurred after their 21st birthday experienced increases in depressive symptoms from the age of 21 to 26 years (
b=1.55, SE=0.66,
t=2.35,
P=0.02 among s/s homozygotes and
b=1.25, SE=0.34,
t=3.66,
P<0.001 among s/l heterozygotes) whereas l/l homozygotes did not (
b=0.17, SE=0.41,
t=0.41,
P=0.68).
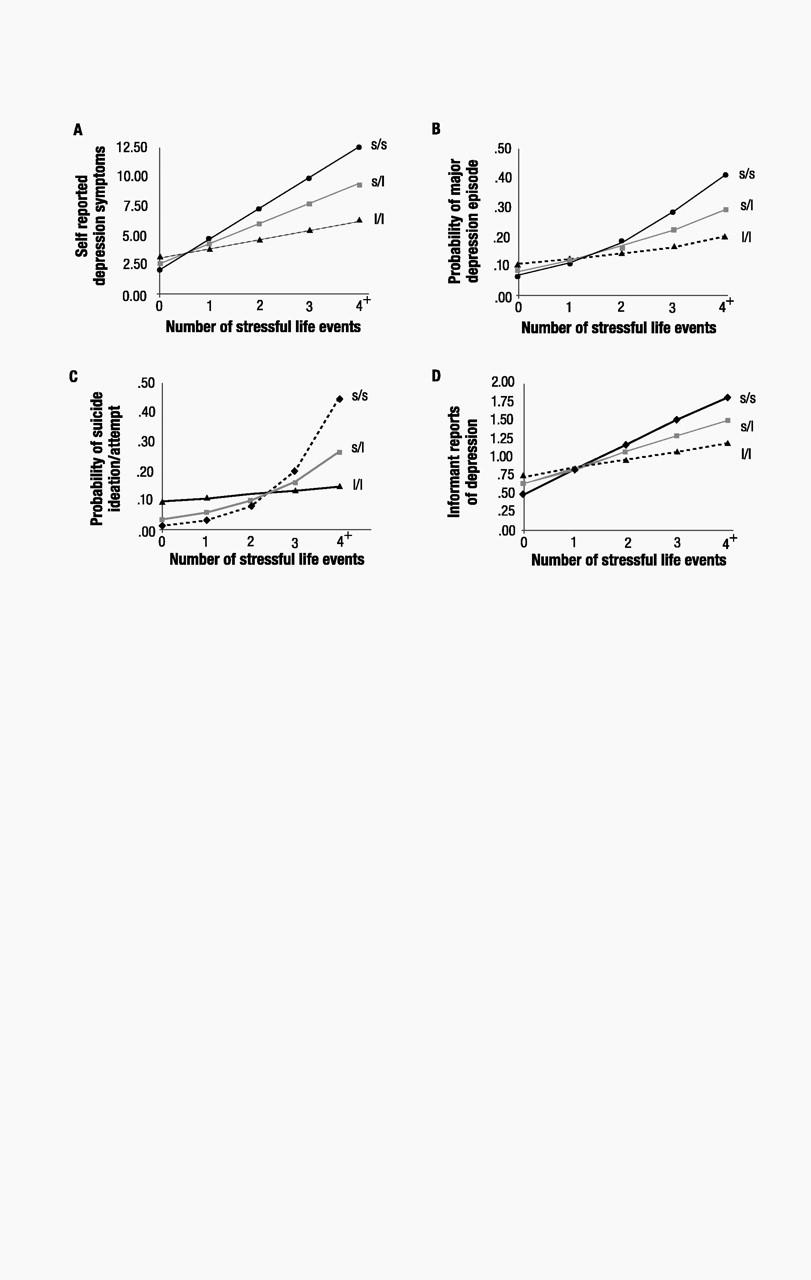
The G × E interaction also showed that stressful life events predicted a diagnosis of major depression among carriers of an s allele but not among l/l homozygotes (P=0.056, Fig. 1B). We further tested whether life events could predict the onset of new diagnosed depression among carriers of an s allele (table S1). We excluded from analysis study members who were diagnosed with depression before age 21. The significant interaction (P=0.02) showed that life events occurring after their 21st birthdays predicted depression at age 26 among carriers of an s allele who did not have a prior history of depression (b=0.79, SE=0.25, z=3.16, P=0.002 among s/s homozygotes and b=0.41, SE=0.12, z=3.29, P=0.001 among s/l heterozygotes) but did not predict onset of new depression among l/l homozygotes (b=0.08, SE=0.20, z=0.42, P=0.67). Further analyses showed that stressful life events predicted suicide ideation or attempt among individuals carrying an s allele but not among l/l homozygotes (P=0.05, Fig. 1C). The hypothesized G × E interaction was also significant when we predicted informant reports of age-26 depression (P<0.01), an analysis that ruled out the possibility of self-report bias (Fig. 1D). The interaction showed that the effect of life events on informant reports of depression was stronger among individuals carrying an s allele than among l/l homozygotes. These analyses attest that the 5-HTT gene interacts with life events to predict depression symptoms, an increase in symptoms, depression diagnoses, new-onset diagnoses, suicidality, and an informant’s report of depressed behavior.
This evidence that 5-HTTLPR variation moderates the effect of life events on depression does not constitute unambiguous evidence of a G × E interaction, because exposure to life events may be influenced by genetic factors; if individuals have a heritable tendency to enter situations where they encounter stressful life events, these events may simply be a genetically saturated marker (
23,
24). Thus, what we have identified as a gene × environment interaction predicting depression could actually reflect a gene × “gene” interaction between the 5-HTTLPR and other genes we did not measure. We reasoned that, if our measure of life events represents merely genetic risk, then life events would interact with 5-HTTLPR even if they occurred after the depression episode. However, if our measure of life events represents environmental stress, then the timing of life events relative to depression must follow cause-effect order and life events that occur after depression should not interact with 5-HTTLPR to postdict depression. We tested this -hypothesis by substituting the age-26 measure of depression with depression assessed in this longitudinal study when study members were 21 and 18 years old, before the occurrence of the measured life events between the ages of 21 and 26 years. Whereas the 5-HTTLPR × life events interaction predicted depression at the age of 26 years, this same interaction did not postdict depression reported at age 21 nor at the age of 18 years (table S2), indicating our finding is a true G × E interaction.
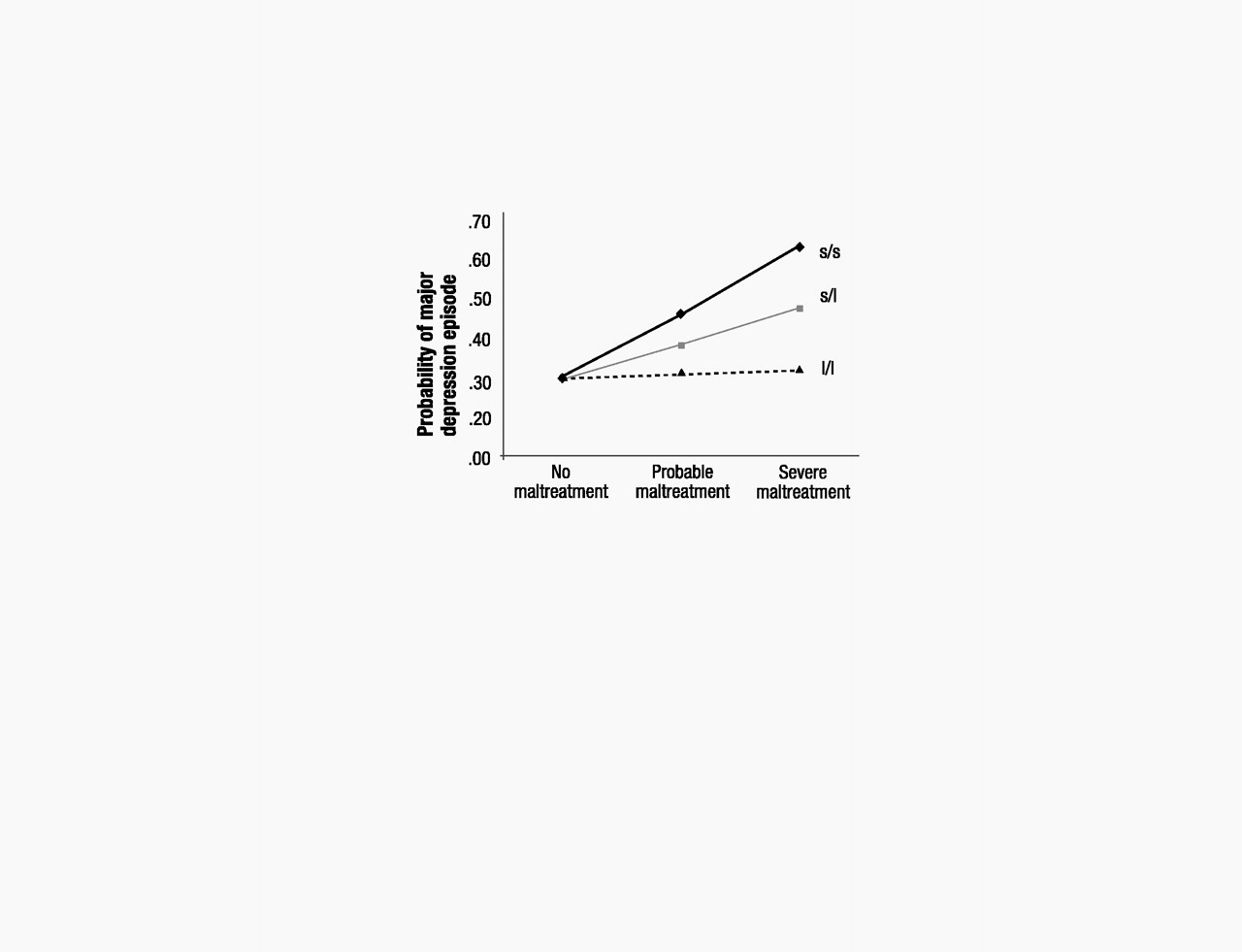
If 5-HTT genotype moderates the depressogenic influence of stressful life events, it should moderate the effect of life events that occurred not just in adulthood but also of stressful experiences that occurred in earlier developmental periods. Based on this hypothesis, we tested whether adult depression was predicted by the interaction between 5-HTTLPR and childhood maltreatment that occurred during the first decade of life (
16,
25). Consistent with the G × E hypothesis, the longitudinal prediction from childhood maltreatment to adult depression was significantly moderated by 5-HTTLPR (table S3). The interaction showed (
P=0.05) that childhood maltreatment predicted adult depression only among individuals carrying an s allele but not among l/l homozygotes (Fig. 2).
We previously showed that variations in the gene encoding the neurotransmitter-metabolizing enzyme monoamine oxidase A (MAOA) moderate children’s sensitivity to maltreatment (
25). MAOA has high affinity for 5-HTT, raising the possibility that the protective effect of the l/l allele on psychiatric morbidity is further augmented by the presence of a genotype conferring high MAOA activity (
13,
26). However, we found that the moderation of life stress on depression was specific to a polymorphism in the 5-HTT gene, because this effect was observed regardless of the individual’s MAOA gene status (tables S4 and S5).
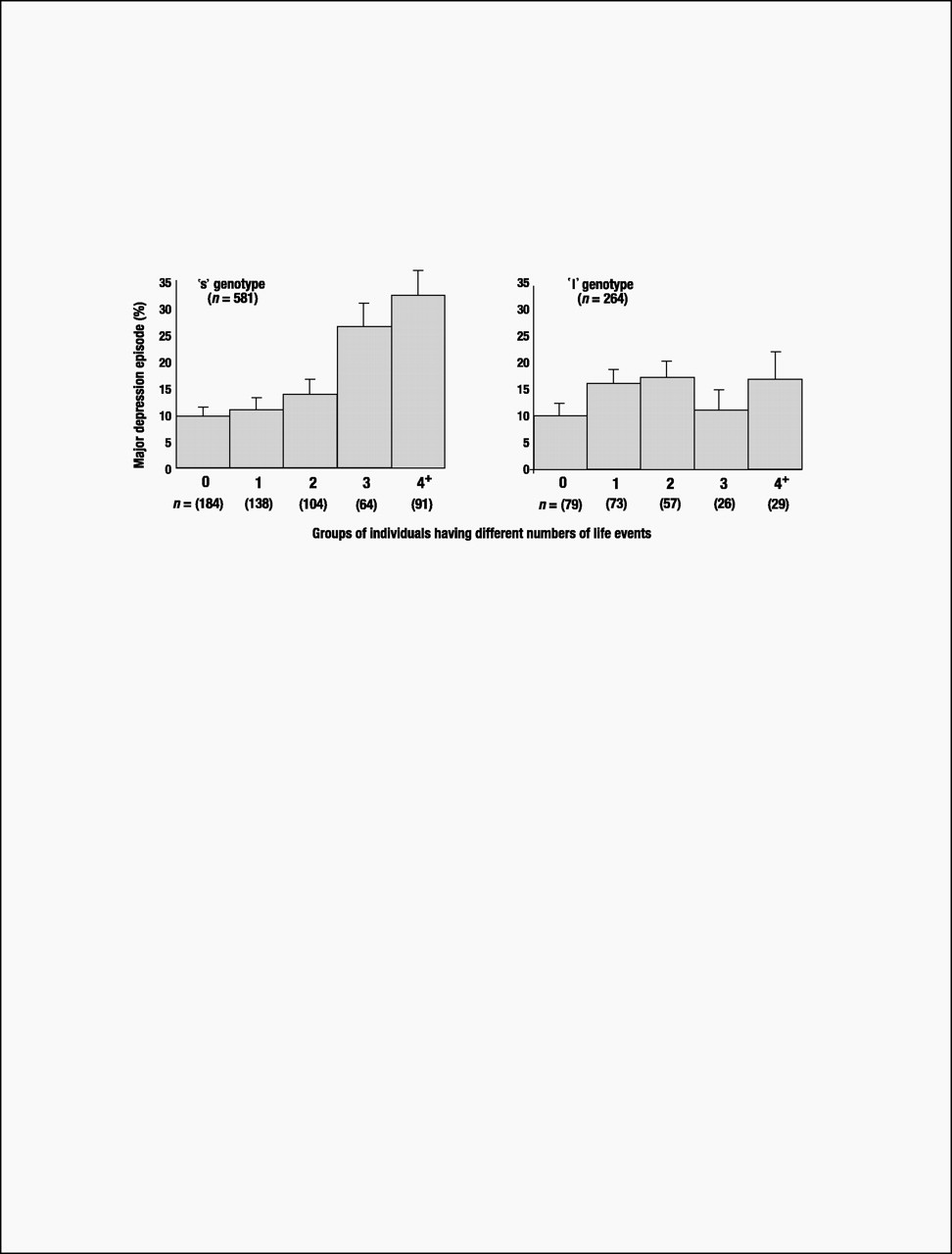
Until this study’s findings are replicated, speculation about clinical implications is premature. Nonetheless, although carriers of an s 5-HTTLPR allele who experienced four or more life events constituted only 10% of the birth cohort, they accounted for almost one-quarter (23%) of the 133 cases of diagnosed depression. Moreover, among cohort members suffering four or more stressful life events, 33% of individuals with an s allele became depressed, whereas only 17% of the l/l homozygotes developed depression (Fig. 3). Thus, the G × E’s attributable risk and predictive sensitivity indicate that more knowledge about the functional properties of the 5-HTT gene may lead to better pharmacological treatments for those already depressed. Although the short 5-HTTLPR variant is too prevalent for discriminatory screening (over half of the Caucasian population has an s allele), a microarray of genes might eventually identify those needing prophylaxis against life’s stressful events (
27).
Evidence of a direct relation between the 5-HTTLPR and depression has been inconsistent (
12), perhaps because prior studies have not considered participants’ stress histories. In this study, no direct association between the 5-HTT gene and depression was observed. Previous experimental paradigms, including 5-HTT knockout mice (
13), stress-reared rhesus macaques (
14), and human functional neuroimaging (
15), have shown that the 5-HTT gene can interact with environmental conditions, although these experiments did not address depression. Our study demonstrates that this G × E interaction extends to the natural development of depression in a representative sample of humans. However, we could not test hypotheses about brain endophenotypes (
28) intermediate between the 5-HTT gene and depression because of the difficulty of taking CSF or functional magnetic resonance imaging measurements in an epidemiological cohort.
Much genetic research has been guided by the assumption that genes cause diseases, but the expectation that direct paths will be found from gene to disease has not proven fruitful for complex psychiatric disorders (
29). Our findings of G × E interaction for the 5-HTT gene and another candidate gene, MAOA (
25), point to a different, evolutionary model. This model assumes that genetic variants maintained at high prevalence in the population probably act to promote organisms’ resistance to environmental pathogens (
30). We extend the concept of environmental pathogens to include traumatic, stressful life experiences and propose that the effects of genes may be uncovered when such pathogens are measured (in naturalistic studies) or manipulated (in experimental studies). To date, few linkage studies detect genes, many candidate gene studies fail consistent replication, and genes that replicate account for little variation in the phenotype (
29). If replicated, our G × E findings will have implications for improving research in psychiatric genetics. Incomplete gene penetrance, a major source of error in linkage pedigrees, can be explained if a gene’s effects are expressed only among family members exposed to environmental risk. If risk exposure differs between samples, candidate genes may fail replication. If risk exposure differs among participants within a sample, genes may account for little variation in the phenotype. We speculate that some multifactorial disorders, instead of resulting from variations in many genes of small effect, may result from variations in fewer genes whose effects are conditional on exposure to environmental risks.